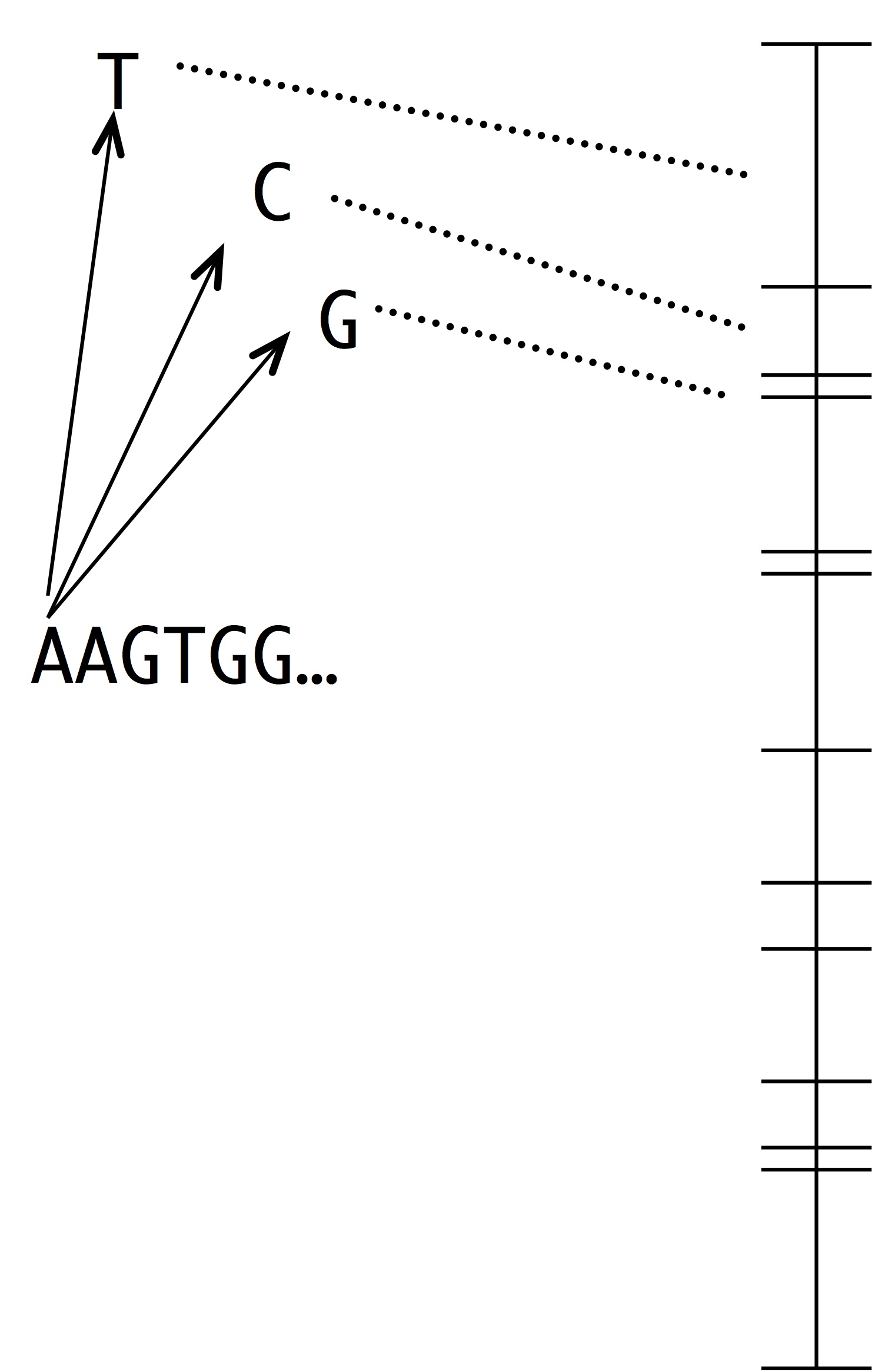
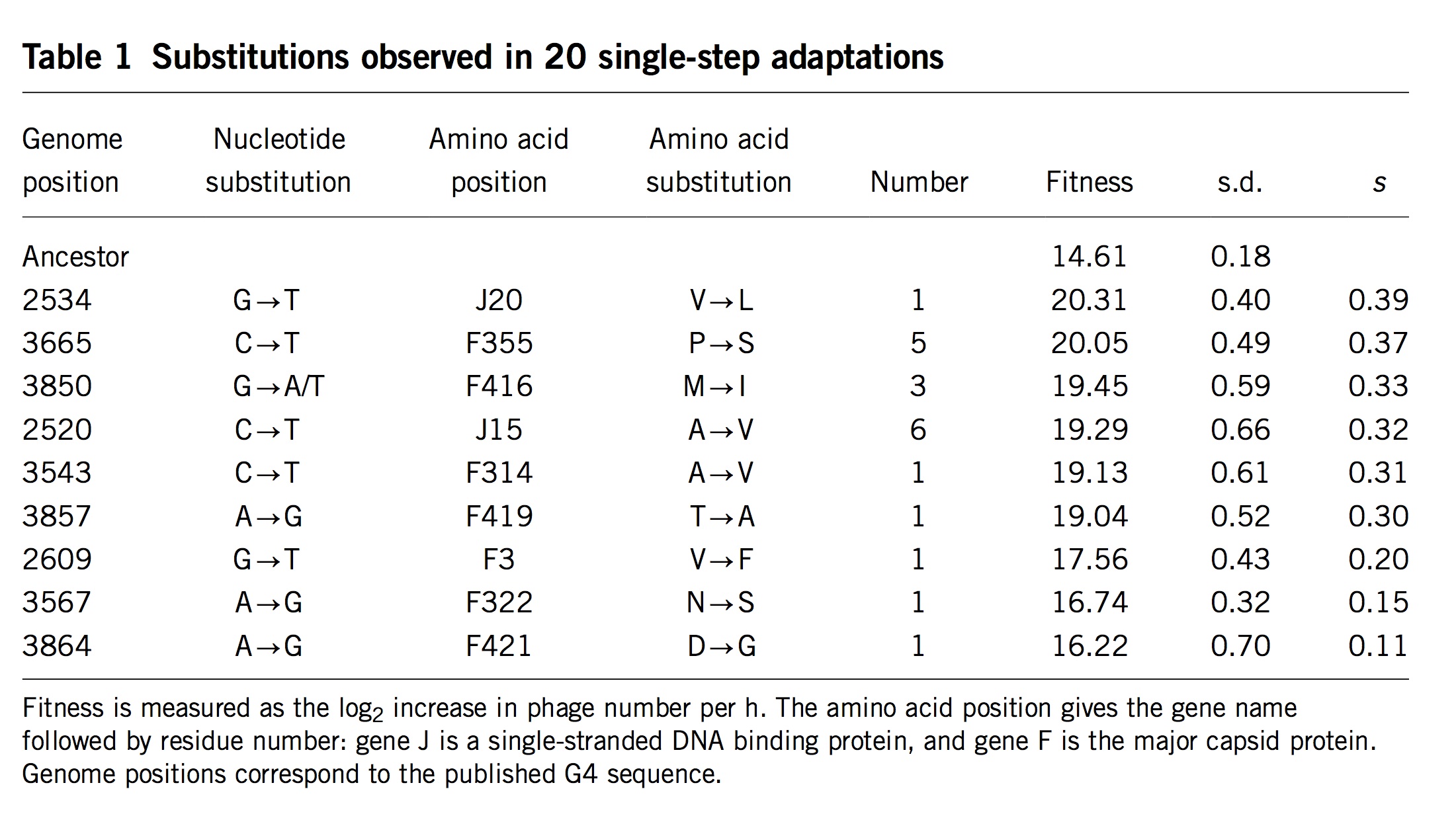
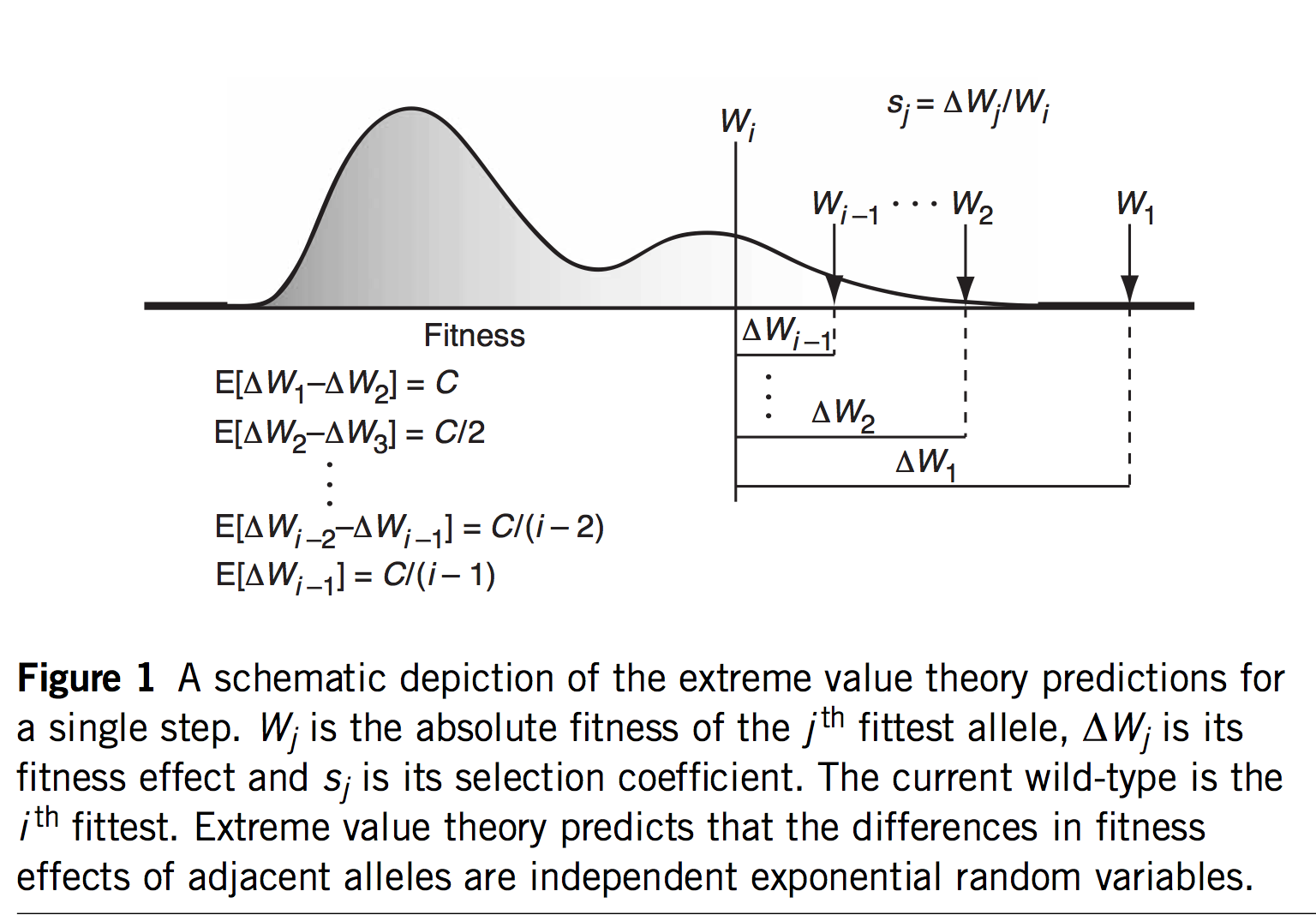
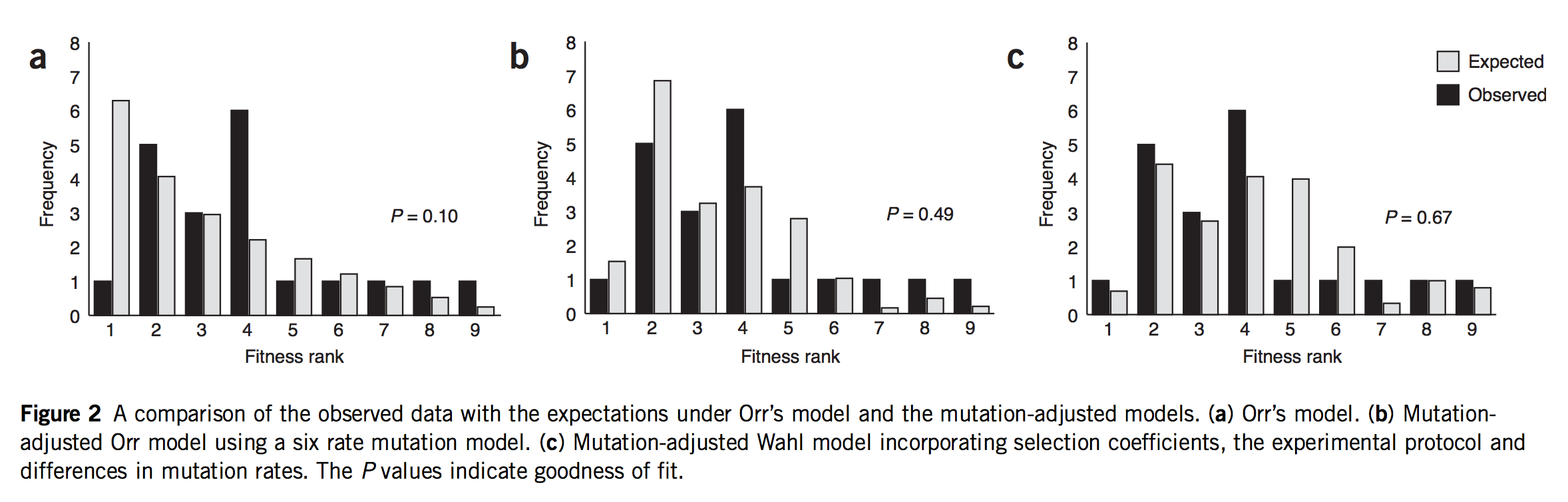
As noted in Bad Takes #3, there is a long tradition of dismissing internalist-structuralist thinking on the false grounds that such ideas are necessarily appeals to teleology or mystical inner urges. If an alternative theory can be dismissed in this manner a priori, as an absurdity, then no further effort is required: no complex theoretical modeling is required, no time-consuming experiments, and no difficult analyses of empirical data. Instead, one simply considers a caricature of the alternative theory, dismisses it as absurd, and goes back to assuming that selection is the ultimate source of meaning and explanation in biology.
Indeed, a key part of the conceptual immune system of the neo-Darwinian thought-collective is a series of facile arguments, each representing some type of excluded-middle or false-dilemma fallacy.
To reject this
Argue against this version
Ignoring this version
Saltation
Evolution only takes leaps; new features must arise fully formed
Evolution normally includes steps or jumps reflecting distinctive variations
Catastrophism
The emergence of key innovations or higher taxa requires catastrophes (revolutions)
Non-normal catastrophes play a disproportionate role in major evolutionary episodes
Mutationism
Evolution is driven by mutation pressure without selection
The timing and character of evolutionary change strongly reflects the timing and character of mutation events
Orthogenesis
Evolution moves in a pre-determined straight line due to mystical inner urges
The course of evolution strongly reflects mutational-developmental channeling of variation
In the long history of evolutionary thinking, one can find examples in which authors advocate, or seem to advocate, ideas in both the second and third columns. If our aim is to identify, explore and evaluate theories about how the world really works, we are naturally drawn to the best version of a theory, not the stupid version most easily knocked down by facile arguments, i.e., a genuine approach to scientific inquiry entails a focus on the third column.
However, if we want to understand how a thought-collective perpetuates itself decade after decade, we need to understand propaganda. Within the neo-Darwinian thought-collective, the focus is on arguments against the theories in the second column, used to reject what Dawkins calls the “doomed rivals” of neo-Darwinism. In Synthesis propaganda, these excluded-middle arguments are used to maintain the all-important TINA doctrine: There Is No Alternative.
In a healthy and diverse intellectual environment, excluded-middle fallacies have no power. However, the Synthesis created an intellectual monoculture. Within this monoculture, the labels for alternative views were repeatedly associated with bad ideas and subjected to ridicule, turning the labels into bogeymen (a bogeyman is an imaginary creature invoked by adults to scare children into compliance). Through ridicule and the fear of ridicule, the system inhibits alternative thinking and preserves the appearance, if not the substance, of a neo-Darwinian status quo. The conceptual immune system is working when gatekeepers use these terms to shut down debate, but the system also works via self-censorship: well meaning scientists, fearing they will be ridiculed as extremists— or merely fearing they will be misunderstood—, avoid using historically correct terminology like “saltation” or “mutationism” or “neo-Darwinism”, and avoid making accurate references to historic debates.
Examples
Orthogenesis appears to be the most successful bogeyman, e.g., the wikipedia page for Orthogenesis repeatedly misleads readers by suggesting that orthogenesis is intrinsically teleological or non-materialistic, in spite of the fact that the article cites, and even directly quotes, multiple scholarly sources that rebut this tendentious linkage. On the talk page, the main author refers bluntly to “God-directed evolution towards a preplanned final goal, which is what orthogenesis is about.” But this is not what orthogenesis is about. Scholarly sources cited in the article (Levit and Olsson; Ulett; Popov; Gould; Schrepfer) repeatedly identify orthogenesis as a general theory about directions that emerge from tendencies of variation, and indicate that the term has been used with a range of views from mystical and teleological to fully materialistic, with well known scientists like Cope or Eimer representing the more respectable materialistic versions. That is, historical scholars describe a general theory, but wikipedians under the influence of Synthesis culture sense that the reason the world needs a wikipedia article on orthogenesis is to serve the conceptual immune system of neo-Darwinism by documenting the foolishness of alternative views.
Self-censorship is well illustrated by scientists who reject gradualism but distance themselves from terms like “saltation” or “macromutation.” For instance, Arthur (2004), after making clear he is a saltationist, e.g., like Goldschmidt, reassures readers he wants to “make clear I am not a ‘saltationist’ like Goldschmidt” (p. 107). Orr and Coyne (1992) utterly ransacked the historic Synthesis case for gradualism yet they say this:
“We hasten to add, however, that we are not ‘macromutationists’ who believe that adaptations are nearly always based on major genes. The neo-Darwinian view could well be correct. It is almost certainly true, however, that some adaptations involve many genes of small effect and others involve major genes. The question we address is, How often does adaptation involve a major gene?”
But under natura non facit saltum, the answer to how often major-effect mutations contribute to adaptation is “almost never,” which (as Orr and Coyne already know) is incorrect. Note that one of the authors of Orr and Coyne (1992) went on to become the go-to defender of orthodoxy for science reporters (see below), and the other eventually went in the opposite direction, citing Bateson approvingly and dismissing gradualism as “little more than a mathematical convenience” (Orr, 2005). Maybe that is why the paper is at war with itself.
Svensson (2023) is deploying the conceptual immune system in the following passage:
Given the strong experimental and empirical evidence against directed mutations (Lenski and Mittler 1993; Futuyma 2017; Svensson and Berger 2019) and the failure of the early mutationists to appreciate the power of natural selection, it is astonishing that some contemporary evolutionary biologists are pushing for a revival of mutationism or mutation-driven evolution (Stoltzfus 2006; Nei 2013; Stoltzfus and Cable 2014). Mutationism was closely connected to the theory of orthogenesis…
Here Svensson encourages readers to join him in scoffing at scientists by linking them to 3 different bogeymen, none of which features in the cited works by Stoltzfus (2006), Nei 2013 and Stoltzfus and Cable (2014).[1] These authors all treat “mutationism” sympathetically, but none rejects the power of selection, e.g., the title of Stoltzfus (2006) is literally “Mutationism and the dual causation of evolutionary change”. None of these pieces invokes directed mutation or advocates what readers would recognize as orthogenesis (i.e., the excluded extreme): Svensson is simply fabricating links to two more bogeymen to increase the chances of ridicule. The piece by Stoltzfus and Cable (2014) is not science but a lengthy historical analysis that debunks the mutationism myth being employed by Svensson in the same sentence.
Importantly, the main clause in the first sentence above from Svensson does not refer to a scientific theory but to persons “pushing for revival.” That is, Svensson does not say that a scientific position is wrong, nor does Svensson express “astonishment” at an idea, but instead the author expresses astonishment at the behavior of persons, i.e., the grammar of the sentence indicates that this is an overt attempt at personal shaming. The “pushing for revival” rhetoric also misrepresents the 3 cited sources, e.g., here is the final paragraph of Stoltzfus (2006):
Nei clearly conceives of his own thinking as “neo-Mutationism”, i.e., something different from historic mutationism, and his main focus is on explaining new thinking and applying it to molecular evolution rather than engaging in historical debates. Stoltzfus and Cable (2014) argue, based on a scholarly analysis of the views of Morgan, Bateson, de Vries and Punnett, that contemporary thinking is closer to their ecumenical view than to neo-Darwinism, i.e., not pushing for revival but reporting on a retreat from a former orthodoxy, one that is easily documented (see The shift to mutationism is documented in our language).
Though the “pushing for revival” rhetoric is false and unscientific, it represents a skillful use of rhetoric by Svensson: he is directly tapping into the conceptual immune system, relying on the fact that his readers have been trained to respond with aversion to the words “mutationism”, “orthogenesis” and “directed mutation.”
Role in Synthesis tribalism
Sometimes, references to the strawman versions of non-Darwinian theories are not focused on ridiculing outsiders or doubters, but on rallying followers to align with their tribal identity. For instance, consider Futuyma (2015):
The seeming exclusivity of the ES [Evolutionary Synthesis] can be understood (and excused, if deemed necessary) only by appreciating the state of evolutionary discourse in the early twentieth century (see Simpson 1944; Rensch 1959; Bowler 1983; Reif et al. 2000). Darwinism was in “eclipse” (Huxley 1942; Bowler 1983), in that almost no biologists accepted natural selection as a significant agent of evolution. (The exceptions were chiefly some of the naturalists.)… Hugo de Vries and Thomas Hunt Morgan, founders of genetics, instead interpreted mutations as a sufficient cause of evolution… [omitted comments on Lamarckism and orthogenesis]… Those who today disparage the Evolutionary Synthesis as a constrained, dogmatic assertion that evolution consists only of natural selection on random genetic mutations within species must recognize that the authors of the Synthesis were responding to an almost complete repudiation of natural selection, adaptation, and coherent connection of macroevolution to these processes.
As an argument about scientific history, this is a chain of fallacies.[1] The first fallacy is that critics of neo-Darwinism such as de Vries and Morgan denied the agency and importance of selection, or that they accepted mutations as a sufficient basis of evolution. But let us suppose that critics of neo-Darwinism denied selection: how does that justify or explain the architects making selection all-important, the exclusive source of order and direction? Wouldn’t it have been wiser to counter the extremists with a more moderate position? That makes two fallacies. Now, set aside those two fallacies, i.e., suppose that critics denied selection and that this somehow justifies advocacy of an opposing extreme. Where does that leave us today? We now have a choice of two extreme theories, neither of which fits the evidence. The logical choice is to declare both of these dinosaurs extinct, right? Given that “excusing” is something we do for people not for theories, why does Futuyma make this a matter of literally “excusing” dead people? And given that we are called on to forgive, why does Futuyma conclude that we must direct out sympathy to one specific group of extremists rather than both?
Of course, the appeal of Futuyma’s rhetoric is based on tribalism, not logic. Synthesis Historiography tells us that, before the Great Synthesis unified the kingdom, bringing a period of peace and prosperity, there was an era of chaos and darkness called The Eclipse, when the light of Darwin was absent, with constant war between the tribes of Lamarckians, Darwinians, mutationists, orthogenesists and saltationists. In order to unify the kingdom and return the throne to the House of Darwin (the rightful rulers), the knights of the Synthesis had to purge the anti-Darwinians, who behave irrationally and hold views with obvious flaws. That is, the origin story for the Synthesis tribe has a historic battle in which the good guys use reason and evidence to beat the bad guys whose heads are full of nonsense.
In the passage quoted above, Futuyma is calling on the power of these shared cultural tropes, naming The Eclipse and all the classic bogeymen— Lamarckism, saltations, orthogenesis and mutationism— to remind tribal members which side they are on: the good guys fighting against the benighted enemies of Darwin and selection. Thus Futuyma skillfully manipulates in-group readers using a shared mythology, with arguments that will seem bizarrely illogical to an outsider.
Capturing the middle ground for Darwin
In most contexts, the system of false-dilemma arguments suffices to maintain ideological conformity and rally the faithful. However, sometimes there are doubters or rebels or merely ordinary scientists who stumble on an unorthodox result and wonder if there might be something of value in alternatives to neo-Darwinism.
In this case, gatekeepers must offer a more sophisticated argument. One common approach is appropriation: conceding some aspect of the alternative view, but describing it in different language, grounding it in familiar sources and associating it with illustrious ancestors, and insisting that this actually part of the mainstream tradition and is not the same thing as any historical alternative to neo-Darwinism.
For instance, when confronted with evidence that evolution may sometimes reflect developmental channeling of variation, the guardians of orthodoxy may admit that the evidence exists, but insist that it is not very compelling, that this is definitely not orthogenesis, and anyway, that this possibility was foreseen by Darwin’s apostles and disciples, therefore it is already part of tribal culture. Futuyma (2017) makes this type of appropriation argument, citing the following passage from Mayr:
“Every group of animals is ‘predisposed’ to vary in certain of its structures, and to be amazingly stable in others . . . Only part of these differences can be explained by the differences in selection pressures to which the organisms are exposed; the remainder are due to the developmental and evolutionary limitation set by the organisms’ genotype and its epigenetic system . . . the epigenotype sets severe limits to the phenotypic expression of such [random] mutations; it restricts the phenotypic potential. The understanding of this limitation facilitates the understanding of evolutionary parallelism and polyphyletic evolution.”
Mayr (1963) p. 608
A better historic source to cite for this idea would have been Eimer or indeed, dozens of other non-Darwinian scientists who advocated much more forcefully for a role of internal developmental biases. However, to cite Eimer would be to go outside the Synthesis tradition.
Mayr typically was disdainful of internalist theories and evo-devo— he said the developmentalists were “hopelessly confused” because they didn’t understand that development is just a proximate cause—, and he repeatedly invoked the contrary neo-Darwinian position that, because natural populations have infinite variability, when the same thing happens twice in evolution, this must be because it is the uniquely apt solution. In the above passage, Mayr appears to have gone off-script (his meaning is not precisely clear: one could interpret this as a statement about epistatic effects mediated by selection). However, Futuyma, (2017) is happy to accept the above statement as justification to fully appropriate developmental bias on behalf of the Synthesis tradition. He literally writes, “The idea that development can influence the direction of evolution was fully congenial to the architects of the ES.” Note that this is a statement about people and not about scientific theories, i.e., there is no explanation of how developmental bias follows from the shifting gene frequencies theory, or why Mayr was wrong to reject developmentalist arguments in the 1980s and 1990s. He does not even attempt such a theoretical reconciliation (for an attempt, see Amundson 2005 or Scholl and Pigliucci 2010). He quotes some people and declares on this basis that the case is closed. He is not defending any theory of causation, but defending the fullness and authority of tradition.
Even as a claim about tradition, the notion that the architects of the Modern Synthesis were “fully congenial” to the idea of internal dispositions is clearly false. For evo-devo people who fought for respectability against Synthesis gatekeepers, this must feel like gaslighting. Mayr is merely stating an idea in vague terms. He doesn’t actually use the key word “direction” in the cited passage, which is important (for those of us who think about these things) because it leaves open whether he is allowing directionally biased effects (more up than down) or merely dimensional effects (more trait 1 than 2). We can’t tell because this is just hand-waving and Mayr has not provided an explicit theory that would clarify such issues. When scientists are serious about an idea, they typically invest intellectual labor in exploring, applying, and defending an idea, clarifying their own thinking, and making important distinctions (e.g., think of how Simpson or Mayr developed terminology for modes of speciation or evolutionary rates). Did Mayr publish any research on internal dispositions? Did he inspire an evo-devo research program with his forceful advocacy of developmental bias? Did he offer a terminology to recognize different classes of bias? No, none of that. A more accurate description of historical sources would be that some of the architects toyed with orthogenesis-adjacent ideas while (more typically) advocating for the standard neo-Darwinian view that, because variation is abundant in all directions, systematic patterns reflect selection and not variation.
Let’s return to the issue of gradualism vs. saltationism. At one extreme is the position of natura non facit salta, i.e., nature does not take leaps. Thinkers such as Fisher and Darwin thought that, for practical purposes, all evolutionary changes are composed from infinitesimal effects. Darwin said that his theory would “absolutely break down” if any organ could not have been formed by a succession of infinitesimal changes. The intermediate position of historic saltationists such as Bateson and T.H. Huxley is that evolution has some jumps. The contrary extreme from gradualism, in which evolution is (for practical purposes) all large jumps, is found only as a strawman theory.
As explained in Why size matters, the gradualist position is not arbitrary for neo-Darwinism and other views that assume empirical adaptationism. If selection and variation are like the potter and the clay, with variation merely providing raw materials and selection providing shape and direction, then variation has to be composed of fine particles. If all change is small, it is possible to argue that selection governs evolution and can do anything, working from infinitesimal variation in every trait. But if evolutionary change comes in chunks, this immediately takes something out of the control of selection—the character and timing of discrete variations—, and then we need some kind of theory for the character and timing of variations in order to have a workable theory of evolution.
In other words, empirical adaptationism necessarily provokes theories of gradualism (in the sense of infinitesimalism). This is not just a logical conclusion, it is how scholars such as Beatty or Gould have reconstructed the actual development of Darwin’s thinking. Likewise, the empirical conclusion that saltations actually occur in evolution provokes the search for internalist theories that address the generation of non-infinitesimal variations. This is not just a logical conclusion, it is why Bateson cataloged distinctive variations in order to study evolution.
Darwin provided some early examples of the excluded-middle arguments outlined above. In his writings, he nearly always embellishes his case against discrete evolutionary steps by referring to them with dramatic language as “monstrosities”:
I reflected much on the chance of favorable monstrosities (i.e., great and sudden variations) arising. I have, of course, no objection to this, indeed it would be a great aid, but I did not allude [in OOS] to the subject for, after much labor, I could find nothing which satisfied me of the probability of such occurrences. There seems to me in almost every case too much, too complex, and too beautiful adaptation, in every structure, to believe in its sudden production.
Why “great and sudden”? Why not “modest”? Why not “medium-sized and sudden”? Describing saltations in provocative and negative language is a common rhetorical trick, e.g., Wright associates them literally with “miracles”:
“From assisting Prof. Castle, I learned at firsthand the efficacy of mass selection in changing permanently a character subject merely to quantitative variability. Because of this and a distaste for miracles in science, I started with full acceptance of Darwin’s contention that evolution depends mainly on quantitative variability rather than on favorable major mutations. ” (Wright S. 1978. The Relation Of Livestock Breeding To Theories Of Evolution. Journal Of Animal Science46:1192-200.)
Note that Wright misrepresents Darwin’s position as calling for “mainly” rather than exclusively quantitative variability, i.e., natura non facit saltum.
This reliance on strawman arguments and misdirection sometimes makes it difficult to determine what Darwin’s followers actually believe. Clearly they are against monstrosities, but how large of a non-monstrosity will they tolerate? Clearly Wright is against miracles and for quantitative variability, but what exactly does that mean? When gradualism fails, will he claim that there are no miracles and insist he was right all along?
Today the issue has been turned on its head. Saltationism is now the mainstream view, but “saltationism” is still presented as the straw-man theory that all evolutionary changes are dramatic leaps, or in which major taxon-defining traits must appear in a single step (e.g., Coyne). As we have seen, the contemporary scientists who conclude in favor of saltationism on empirical or theoretical grounds insist that they are not saltationists, and they sincerely hold the erroneous belief that they are aligned with Darwin and historical neo-Darwinism.
This is what happens when people absorb the TINA doctrine, i.e., they learn the lesson that neo-Darwinism is just what is reasonable, and all the alternatives are crazy, without learning neo-Darwinism as a substantive falsifiable position. Scientists are typically agile thinkers, great at making up rationalizations: if you train them to accept that gradualism is right, they will find some way to make it right, based on whatever assumptions and definitions yield the approved conclusion. For instance, note how the issue is framed by researchers cited by Chouard:
Many researchers have welcomed the return to favour of large-effect mutations and have resurrected Goldschmidt’s long reviled idea of the hopeful monster. But they can’t ignore the small-effect mutations. “We need much more data before the issue of large versus small can be settled”, says Coyne. Kingsley, like Coyne favours a middle-ground view, in which neither large- nor small-effect mutations are ruled out. “Our work has too often been portrayed as saying that Darwin was wrong” about big leaps in adaptation, he says. But in fact, none of the traits his group has studied is completely due to the effects of a single gene.
Instead of defending the all-small position of genuine neo-Darwinian gradualism, the traditionalists now defend the not-all-large (i.e., some-small) position that one “can’t ignore the small-effect mutations.”
Likewise, in response to Shapiro’s criticism that molecular saltations speak against Darwinism, Dean (2012) objects thus:
His stance is patently unfair. Thomas Huxley famously criticized Darwin for championing too gradualist a view of phenotypic evolution. Today’s Darwinists accept Huxley’s criticism . . . Horizontal gene transfer, symbiotic genome fusions, massive genome restructuring (to remarkably little phenotypic effect in day lilies and muntjac deer), and dramatic phenotypic changes based on only a few amino acid replacements are just some of the supposedly non-Darwinian phenomena routinely studied by Darwinists.
Notice the charge of being “unfair.” Here the author has gone all the way to a purely cultural position on neo-Darwinism: there is no fixed scientific theory attached to the brand, only a cultural tradition consisting of people (Darwinians) whose beliefs may change at any time. If the right sort of people start studying saltations or invoking them, this makes saltations part of neo-Darwinism.
Conclusion
I could go on, but these examples should be sufficient to make the point about how the conceptual immune system works. Excluded-middle arguments based on ridicule are the first line of defense, but when scientists stumble upon the middle ground, traditionalists will claim it for tradition, even if this involves misrepresenting history and shifting the goal-posts. When apologists for tradition shift from merely rejecting strawmen to appropriating the excluded middle on behalf of tradition, this represents a genuine scientific shift that is masked by conservative rhetoric. Being able to see through the misleading rhetoric is a skill that can be learned. In the current climate of evolutionary discourse, it is a necessary skill.
Another necessary skill is courage. The secret power of the conceptual immune system is that the excluded-middle arguments rely on ridicule delivered by gatekeepers, and the aura of ridicule remains when scientists discover the excluded middle.[2] Scientists want to be respected, not ridiculed, by their peers: this threat is enough to cow most of them into voluntarily making the wrong association, in order to avoid disrespect.If you use terms like “saltation”, “orthogenesis” or “mutationism” positively (or invoke “neo-Darwinism” negatively) even if your usage is historically correct and accurate, this guarantees that you will be subjected to knee-jerk reactions of ridicule, or you will be accused of seeking attention using inflammatory rhetoric. Clearly the past generation of scientists was afraid of saying such words even when they are perfectly apt and reasonable.[3] I hope that current and future generations will not be so fearful.
References
Arthur W. 2004. Biased Embryos and Evolution. Cambridge: Cambridge University Press.
Dean (2012) Review of Evolution: a View from the 21st Century. Microbe Magazine (available via the wayback)
Gould SJ. 2002. The Structure of Evolutionary Theory. Cambridge, Massachusetts: Harvard University Press.
Levit GS, Olsson L. 2006. “Evolution on Rails”: Mechanisms and Levels of Orthogenesis. Annals for the History and Philosophy of Biology 11: 97-136.
Mayr E. 1963. Animal Species and Evolution. Cambridge, Massachusetts: Harvard University Press.
Orr HA, Coyne JA. 1992. The Genetics of Adaptation: A Reassessment. American Naturalist 140:725-742.
Popov I. 2009. The problem of constraints on variation, from Darwin to the present. Ludus Vitalis 17:201-220.
Ulett MA. 2014. Making the case for orthogenesis: The popularization of definitely directed evolution (1890–1926). Studies in History and Philosophy of Science Part C: Studies in History and Philosophy of Biological and Biomedical Sciences 45:124-132.
Notes
[1] For a takedown of this passage from Futuyma, see Stoltzfus (2017). Critics of neo-Darwinism typically objected to its two main vulnerabilities, natura non facit saltum and the dichotomy of roles in which selection is the potter and variation is the clay. In every alternative to neo-Darwinism, the variation-generating process plays a dispositional role. Note that, in attempting to understand what critics of neo-Darwinism believe, one must not mistake skepticism about the quality and rigor of selective explanations for doubts about the (in principle) power of selection. Bateson did not deny selection as a true cause responsible for adaptation, but he was so jaded he thought that selective explanations would always be inaccessible to scientific proof and would remain mere armchair speculation. Nei seems to have a similar position.
[2] Because ridicule is such a strong demotivator, maintaining conformity does not require a large class of aggressive gatekeepers a la Svensson. A little bit goes a long way. As explained in the introductory paragraphs, most people don’t need to be shamed directly to avoid thinking unorthodox thoughts, they will just do it on their own because, again, people want to be respected.
[3] Note that accurately depicting mutationism, neo-Darwinism, or other historic views does not require any special courage for historians or other scholars who are not striving for, and are not dependent on, the approval of mainstream Synthesis culture. Among mainstream evolutionary thinkers, Allen Orr has shown courage in quoting Bateson approvingly and in depicting gradualism correctly as an extreme position. Gould was unafraid to show sympathy to non-Darwinian thinking, but his sympathy seems more like pity when one notices how frequently Gould associates critics of neo-Darwinism (e.g., de Vries, Goldschmidt, Bateson) with behavioral disorders, reinforcing the Synthesis tribal mythology in which only crazy people reject neo-Darwinism.
Unfamiliar ideas are often mis-identified and mis-characterized. It takes time for a new idea to be sufficiently familiar that it can be debated meaningfully. We look forward to those more meaningful debates. Until then, fending off bad takes is the order of the day! See the Bad Takes Index.
In his equally entertaining and obnoxious piece “The frailty of adaptive hypotheses for the origins of organismal complexity,” Lynch (2007) writes
The notion that mutation pressure can be a driving force in evolution is not new (6, 24–31)
citing works of Darwin, Morgan, Dover, Nei, Cavalier-Smith, and Stoltzfus and Yampolsky.
What does it mean to invoke evolutionary change due to a driving force of mutation pressure? This language suggests a process of population transformation by the mutational conversion of individuals, in contrast to population transformation by reproductive replacement.
That is, in a simplified world of discretely inherited types, we can imagine two general ways to transform a reproducing population from mainly type A to mainly type B. One mode is for an initially rare type B to take over the A population, over many generations, by the cumulative effects of differential reproduction, either biased (selection) or unbiased (drift). Individuals of type B over-produce, while A individuals die out, so that A individuals are replaced by unrelated B individuals. This is usually how we think about the transformation of populations: reproductive replacement. Selection and drift are often listed as the two main causes of evolution, and they act by reproductive replacement.
A second possible mode of change is for a population of predominantly A individuals to change by many separate events of A-to-B conversion (either individual conversion, or the cross-generational conversion of a lineage from parent to offspring). In this case, A individuals are lost, not by death, but by conversion, and likewise, B individuals are over-produced, not by the excess reproduction of B parents, but by conversion from A individuals. This process might take a single generation or many generations, depending on the rate of conversion (see image for a simulation).
A simulation of evolution by mutation pressure (from the mutation pressure page developed by John McDonald). N = 20 red squares at left represent individuals, with offspring generations going to the right. The reproductive variance is 0, so each individual leaves exactly 1 offspring in the next generation, inheriting the parental state or a mutated state with u = 0.025. By 100 generations, the population is mostly transformed, by mutational conversion alone, without reproductive differences.
In a more complex scenario, there are other possibilities. For instance, given diploid inheritance we could consider a process of biased gene conversion by which A1A2 genotypes are converted into A2A2. Suppose that A2 is recessive so that A1A1 and A1A2 have phenotype P1, and A2A2 has P2. In this scenario, biased gene conversion can transform a predominantly P1 population into a P2 population. Dover’s ideas about molecular drive combine effects of conversion and replacement.
One of the minor theories in Darwin’s Origin of Species is the mass transformation of individuals by direct effects of the environment. This idea was not unique to Darwin, but simply reflected 19th-century thinking by which heredity is (in effect) mediated by responsive memory-fluids that circulate in the body: after collecting bodily experiences, the memory-fluids gather in the gametes, and during reproduction, they blend, passing on a blended version of inheritance plus experience. Given this view, it was natural to suppose that, when animals or plants encounter a new environment, this results in a hereditary transformation by the cumulative effect of many environment-induced conversions.
The literature of the pre-Synthesis period includes some (typically ambiguous) references to population transformation by mutational conversion, e.g., Shull (1936) writes
If a given mutation were to happen often enough, and nothing opposed its survival, it could easily spread through the entire species, replacing all the other genes at the same locus.
Evolution by mutation pressure according to Haldane and Fisher
In the broader context of evolutionary theorizing, the mutation pressure theory appears most prominently as a strawman rejected by Haldane (1927, 1932) and Fisher (1930). That is, Haldane and Fisher did not advocate the notion of evolution by mutation pressure, but presented an unworkable theory as a way to reject the idea, popular among critics of neo-Darwinism, that evolutionary tendencies may reflect internal variational tendencies. In reality, the early geneticists typically argued, not for mutational transformation of populations, but for a two-step process of “mutation proposes, selection disposes” (decides); and the idea of orthogenesis was typically an appeal to what we might call “constraints” today. That is, the mutation pressure theory began as a dubious take on internalist thinking.
Regardless, Haldane and Fisher worked out the implications of evolution by mutation pressure, finding it unlikely on the grounds that, because mutation rates are small, mutation is a weak pressure on allele frequencies, easily overcome by opposing selection. Haldane concluded that this pressure would not be important except in the case of neutral characters or abnormally high mutation rates.
The conclusion of Haldane (1927)
To understand what Haldane is doing, one must bear in mind that, in the neo-Darwinian tradition, selection is the model of an evolutionary cause: other factors or processes are considered to be causal only to the extent that they look like selection. What selection does is to shift frequencies (and ultimately drive alleles to fixation), so Darwin’s modern followers define evolution as shifting frequencies and they define causal forces as pressures that might cause fixation. In effect, Haldane equates the importance of mutation with the potential for mutation pressure to drive allele frequencies. In this way of thinking, if mutation-biased evolution is happening, this is because mutation is driving alleles to high frequency against the opposing pressure of selection, which leads to Haldane’s conclusion that either (1) the mutation rate has to be abnormally high, or (2) selection has to be practically absent (i.e., neutrality). Fisher’s (1930) reasoning was similar. From the observed smallness of mutation rates, he drew a sweeping conclusion to the effect that internalist theories are incompatible with population genetics.
Provine (1978) identifies this argument (against evolution by mutation pressure) as one of the key contributions of theoretical population genetics to the Modern Synthesis, because it gave Darwin’s followers a seemingly rigorous basis to reject internalist theories (establishing the core Synthesis principle of There Is No Alternative). The argument was cited repeatedly by the architects of the Modern Synthesis (for examples, see Stoltzfus, 2017), and continues to be cited, e.g., Gould (2002) cites Fisher’s version of the argument and concludes that
Since orthogenesis can only operate when mutation pressure becomes high enough to act as an agent of evolutionary change, empirical data on low mutation rates sound the death-knell of internalism. (p. 510)
Subsequent work has partially undermined the narrow implications of the Haldane-Fisher argument, and completely undermined its broader application as a cudgel against internalism. Mutation pressure is almost never a reasonable cause of population transformation, because it would happen so slowly and take so long that other factors such as drift would intervene, as argued by Kimura (1980). The case studied by Masel and Maughan (2007) is a rare example in which evolution by mutation pressure is reasonable: the authors estimate an aggregate mutation rate of 0.003 for loss of a trait (sporulation) dependent on many loci, concluding that complex traits can be lost in a reasonable period of time due primarily to mutational degradation.
Thus, in spite of what one would conclude from Haldane (1927), patterns of mutation bias in evolution generally do not indicate evolution by mutation pressure via high mutation rates, or via neutral characters. Mutation-biased neutral evolution happens, not because mutation pressure is driving alleles to fixation in a biased way (instead, drift is the cause of fixation), but due to a bias in the origination process. And of course, Yampolsky and Stoltzfus (2001) showed that, when there is a bias in the introduction process, this can impose a bias on the course of evolutionary change even when fixations are selective, i.e., there is no requirement for neutral evolution.
In summary, the classic theory of evolution by mutation pressure is not much use in understanding evolution, and is mainly of historical interest for its role in an influential fallacy: generations of evolutionary thinkers believed wrongly that the mutation pressure theory proves mathematically that internalist theories are incompatible with population genetics.
Other theories
Now, with this background, we may return to Lynch’s bad take, associating various authors with mutation pressure as a driving force. Of the authors cited — Darwin, Morgan, Dover, Nei, Cavalier-Smith and my colleagues and I — none of them directly propose a theory of evolution by mutation pressure. However, the ideas of Darwin and Dover depict a process reliant on mass conversion: in Dover’s case, population transformation takes place by a dual process of conversion (gene conversion or sub-genomic replication) and reproductive replacement, and in Darwin’s case, it takes place by direct inherited effects of the environment.
Nei refers to “mutation-driven” evolution (the title of his 2013 book), but this is not a reference to mutation driving alleles to fixation. Nei’s usage of “drive” is descriptive or explanatory: evolution is mutation-driven to the extent that our understanding of important aspects of the course of evolution relies on knowing which mutations happen at what times. The same meaning is used in “Mutation-Driven Parallel Evolution During Viral Adaptation” (Sackman, et al. 2017). For an explanation of this meaning of “drive,” see Bad Take #4.
Likewise, the work from my colleagues and me is not about evolution by mutation pressure. From the very beginning, we have (1) followed Provine (1978) in noting the historical importance of the Haldane-Fisher argument against evolution by mutation pressure, and (2) promoted a theory for the effects of biases in the introduction process, obviously a different theory because it contradicts the implications of the mutation pressure theory.
So, what on earth does Lynch mean when he refers to evolution driven by mutation pressure? This is unclear. The model that Lynch (2007) presents immediately after the quoted statement is not a model of evolution by mutation pressure in the classic sense of Haldane and Fisher and IMHO does not correspond to what any of the cited authors are trying to say.
To understand what the model tells us, we must analyze it in detail, in comparison to the classic mutation-selection balance (also co-developed by Haldane and Fisher). The forces of population genetics are conceptualized like the laws of statistical physics, as mass-action pressures on allele frequencies due to the aggregate effect of countless individual events. In the case of mutation, countless individual events of mutational conversion from allele A1 to allele A2 result in a force or pressure of mutation shifting quantities of A1 to A2. Because there are innumerable independent events, each with an infinitesimal effect, we can represent the aggregate effect with a continuous quantity, e.g., we can write fA2‘ = fA2 + u fA1 to indicate the increase in fA2 due to mutation at rate u from allele A1, and we can write fA1‘ = (1 – u) fA1 to represent the corresponding reduction in the frequency of allele A1 due to mutation to allele A2.
In the classic conception of the mutation-selection balance, if A1 is favored over A2 by a selection coefficient s, then reproductive replacement by selection represents a pressure of magnitude s increasing fA1 and decreasing fA2, whereas mutation is a pressure of magnitude u with the opposite effect, acting by conversion (rather than reproductive replacement). The equilibrium frequency of A2 is roughly f = u / s, and this is typically a small number (much closer to 0 than to 1) because mutation rates are very small, e.g., a typical rate for a specific nucleotide mutation is 10-9 per generation. This is why Haldane concluded (above) that mutation would be unimportant unless selection is effectively absent (i.e., neutrality) or mutation rates are abnormally large (note how the classical mutation-selection balance of Haldane and Fisher is closely related to their argument about evolution by mutation pressure).
Lynch appears to reach a different solution to the same problem of the equilibrium frequency in a 2-allele system. His equation for the ratio of A1 to A2 is meS, where m is the forward-backward mutation bias favoring A1, and eS is the ratio of fixation probabilities where upper-case S = 2Ngs and lower-case s has the same meaning as above. However, this result actually does not represent the equilibrium frequency of A1 in a population of individuals, as for Haldane-Fisher: instead, it refers to the equilibrium distribution of infinitely many loci subject to an origin-fixation process, where each locus is fixed for A1 or A2, that is, meS is the expected ratio of (1) the fraction of loci fixed for A1 to (2) the fraction of loci fixed for A2.
(Figure 1 of Lynch, 2007)
This is easier to understand with a concrete example: the relative genomic frequency of two synonymous codons like CAT and CAC encoding histidine, where one of the codons (let’s assume CAC) is slightly more favored by selection.
The two cases, classic and Lynch, correspond to the large-population and small-population approximations for the ratio of favored to disfavored codons in Bulmer’s (1991) mutation-selection-drift model. In a large deterministic population, each histidine codon is fixed for the favored synonym (CAC), yet the disfavored codon (CAT) is maintained at low frequency in mutation-selection balance per Haldane-Fisher. In the small population, each histidine codon is fixed for either the favored codon (CAC) or the disfavored codon (CAT), and the frequency distribution of fixed states for loci is determined by the balance of two origin-fixation rates. So, in either case, if the disfavored codon is expected a fraction f of the time, then f is also the expected frequency of that codon over an infinite genomic set of histidine sites.
Thus, Lynch’s argument gives a different result because it refers to a different kind of mass-action pressure than Haldane and Fisher conceived. The relevant pressure in Lynch’s argument is the mass-action pressure due to events of origination aggregated over an infinite distribution of loci (sites). This origination pressure is not the same as classic mutation pressure, which is the mass-action pressure due to mutational conversion events aggregated over infinitely many alleles (in a population) at the same locus.
The result of this pressure (relative to a deterministic universe with only the favored codon), is to ensure that, for small values of S = 2Ngs, a substantial fraction of loci are fixed for the disfavored state; when S = 2Ngs becomes modestly large, this fraction is negligible. That is, mutation pressure, for Lynch, refers to something that ensures the predictable presence of deleterious states. By contrast, the theory of Yampolsky and Stoltzfus (2001) is about the way that biases in origination impose biases on which path, out of many possible, is taken by adaptation.
Clearly this model ensures the presence of deleterious states for small values of S, but it is not clear what justifies Lynch’s framing of this as an effect of a pressure of mutation (more precisely, a pressure of origination), rather than as an effect of random drift or of origin-fixation pressure. Mutation and fixation do not act separately in the context of the argument, and drift is profoundly important in ensuring that, in Lynch’s stochastic anti-paradise, a substantial fraction of everything is in a sub-optimal state. In a world that has deterministic selection, the favored codon always wins (and the disfavored one is never fixed by chance), even in small populations, and this will be true regardless of what we assume about mutation. Metaphorically, mutation is just knocking at the door, offering bad choices: drift has to open the door and let them in. On this basis, Lynch ought to point the finger at drift (not mutation) as the reason for non-optimality.
In fact, this is all utterly misleading when taken in context. After telling the reader that the idea of evolution by mutation pressure is not new, Lynch continues as follows
The notion that mutation pressure can be a driving force in evolution is not new (6, 24–31), and the conditions that must be fulfilled if mutation is to alter the direction of evolution relative to adaptive expectations are readily derived.
This is a sweeping claim about mutation and directionality! Yet, what follows is not a general model of effects of mutation on the direction of evolutionary change, but Bulmer’s model featuring fixations of deleterious alleles! That is, Lynch refers generally to mutational effects altering the “direction” of evolution, yet apparently, given his Manichean worldview, “direction” is just a matter of down vs up in fitness. And we just established that drift, not mutation, is the cause of deleterious fixations, which will happen even if there is no mutation bias (e.g., the disfavored codon will sometimes be fixed, even without any mutation bias). In the end, Lynch has presented correct mathematical results, but framed these results in an incorrect way that can only lead to confusion.
Accordingly, Svensson (here or here) has repeatedly claimed, citing Lynch’s paper, that an effect of mutation bias on adaptation would require “drift in small populations.” This error arises from a literal reading of Lynch, who (1) conflates diverse ideas (including ours) under the heading of evolution by mutation pressure, and then (2) makes a sweeping reference to mutational effects on “direction.” However, as explained, the effect requiring small populations in Lynch’s model is the fixation of a slightly deleterious allele by drift in small populations, whereas arguments about mutation-biased adaptation do not involve fixations of deleterious alleles at all, e.g., the behavior of the Yampolsky-Stoltzfus model does not rely in any sense on the fixation of deleterious alleles by drift.
What is the cause of so much misapprehension? The molecular revolution induced profound changes in thinking that have not been properly processed. Instead, we have attempted to squeeze a new understanding into the same old vocabulary— using old words for new concepts. In some cases, the result is verbal violence, as in the way that “Darwinian adaptation” is now used for the lucky mutant view previously known as a non-Darwinian theory of pre-adaptation. Familiar words are now overloaded with different concepts, and we have not paid attention to the problems caused by this overloading. Evolution by mutation pressure, in the classic Haldane-Fisher sense, means something different than what Lynch’s model means, which is something different than what the Yampolsky-Stoltzfus model means. The forces theory is inadequate, and leads scientists to incorrect conclusions, e.g., the assumption that mutation-biased evolution requires neutrality, which is pervasive in the literature.
The path toward greater clarity depends on making distinctions, e.g., distinguishing the introduction (origination) process from classical mutation pressure (across infinitely many copies of an allele in a population) and from origin pressure (across infinitely many loci). The reason to distinguish these, again, is that they behave differently, so that the rules for reasoning about one kind of causal process are different from the rules for reasoning about another.
Likewise, one must bear in mind that biological processes are not the same as the operators in models or mathematical formalisms, which capture only some of implications of biological processes for evolution. The classic conception of forces in population genetics includes a thing with the label “mutation” (and another thing with the label “selection”), but this thing does not have all the same implications as the biological process with the label “mutation.”
Sources that fail to make such distinctions will mislead readers with the impression that every reference to mutation is a reference to exactly the same evolutionary theory, when this clearly is not the case.
References
Fisher RA. 1930. The Genetical Theory of Natural Selection. London: Oxford University Press.
Gould SJ. 2002. The Structure of Evolutionary Theory. Cambridge, Massachusetts: Harvard University Press.
Haldane JBS. 1927. A mathematical theory of natural and artificial selection. V. Selection and mutation. Proc. Cam. Phil. Soc. 26:220-230.
Haldane JBS. 1932. The Causes of Evolution. New York: Longmans, Green and Co.
Haldane JBS. 1933. The part played by recurrent mutation in evolution. Am. Nat. 67:5-19.
Kimura M. 1980. Average time until fixation of a mutant allele in a finite population under continued mutation pressure: Studies by analytical, numerical, and pseudo-sampling methods. Proc Natl Acad Sci U S A 77:522-526.
Lynch M. 2007. The frailty of adaptive hypotheses for the origins of organismal complexity. Proc Natl Acad Sci U S A 104 Suppl 1:8597-8604.
Provine WB. 1978. The role of mathematical population geneticists in the evolutionary synthesis of the 1930s and 1940s. Stud Hist Biol. 2:167-192.
Shull AF. 1936. Evolution. New York: McGraw-Hill.
Stoltzfus A. 2006. Mutationism and the Dual Causation of Evolutionary Change. Evol Dev 8:304-317.
Yampolsky LY, Stoltzfus A. 2001. Bias in the introduction of variation as an orienting factor in evolution. Evol Dev 3:73-83.
Notes
Biased gene conversion is a newly recognized population-genetic force, non-identical with mutation, selection, or recombination. BGC is a conversion mechanism, not a replacement mechanism, but the formula for BGC is A1 + A2 –> A2 + A2, whereas the mutation formula is A1 –> A2. Similarly, the crossing-over formula is A1B1 + A2B2 –> A1B2 + A2B1. Thus, although the molecular operation of gene conversion is associated with cross-overs and with the machinery for recombination, the genetic operation of BGC is not the same thing as recombination.
Unfamiliar ideas are often mis-identified and mis-characterized. It takes time for a new idea to be sufficiently familiar that it can be debated meaningfully. We look forward to those more meaningful debates. Until then, fending off bad takes is the order of the day! See the Bad Takes Index.
In regard to reports of mutational biases influencing the changes involved in molecular adaptation, Svensson and Berger (2019) write
Despite the importance of mutations in these two studies, we emphasize that selection ultimately drove these adaptive allele frequency changes, rather than evolution being ‘mutation-driven’ as some might claim [1,7,8,13].
Actually the “mutation-driven” language is advocated in reference #1 (Nei’s book), but not in the other 3 sources cited, which are Yampolsky and Stoltzfus ( 2001), Stoltzfus (2006) and Stoltzfus and Cable (2014).
The authors object that, whereas the term “drive” refers to a cause that drives an allele to fixation, the changes implicated in the cited studies reflect selective fixation rather than fixation by mutation. The implication is that sources 1, 7, 8 and 13 advocate a theory of population transformation, not by reproductive replacement (via selection or drift), but by mutation pressure, i.e., the cumulative effect of many events of mutational conversion, which is generally a bad idea for reasons pointed out by Kimura (1980), although there are cases where it makes sense, e.g., loss of a complex character (for a more thorough explanation, see Bad take #2).
But of course, fixation by mutation pressure is not the theory advocated in Nei’s 2013 book Mutation-Driven Evolution, nor the other sources cited, nor sources such as this (note the title):
Nor is this what Pennings, et al. (2022) mean when they clarify that “our study is focused on the dynamics of adaptation and reversal in the context of point mutation-driven, stepwise evolution, rather than evolution through horizontal gene transfer or plasmid conjugation”. Nor is this what Tenaillon (2014) means when he writes
“In particular, the long-term evolution of 12 replicate populations of Escherichia coli by R.E. Lenski unraveled a succession of mutation fixations that reached up to 10 % fitness effect (Lenski & Travisano 1994). Large effect mutations appeared therefore to be the drivers of adaptation.”
Furthermore, Svensson and Berger (2019) surely know that authors such as ourselves or Masatoshi Nei, a famous population geneticist, are not advocating fixation by mutation pressure rather than by selection (or drift). For instance, the equation from Yampolsky and Stoltzfus that they recreate in Box 1 is based explicitly on the probability of fixation for a beneficial allele given by Haldane (1927).
That is, Svensson and Berger are making what is called a “bad-faith argument”, an argument that they know is wrong but which they use anyway, trusting that the argument gain favor with naive readers.
One must remember that the piece by Svensson and Berger (2019) is not a serious scholarly analysis, but a parody of Bad Synthesis Apologetics, a Sokal’s hoax exposing that— when the topic is either post-modernist cultural analysis or the status of evolutionary theory— it is possible to “publish an article liberally salted with nonsense if (a) it sounded good and (b) it flattered the editors’ ideological preconceptions.”
With their cheeky “mutation-driven” objection, the authors are parodying the kind of bad-faith argument that does not address any genuine issue of dispute, but is simply a way to score points with the kind of guileless reader who thinks Masatoshi Nei needs a lesson in basic population genetics from Svensson and Berger. It is a long-standing part of Synthesis culture to believe that critics of orthodoxy behave irrationally and hold views with obvious flaws.
If we take away this false pretense, the remaining issue is semantic: (1) does “mutation-driven” refer distinctively to the case in which mutation is a cause of allele fixation, i.e., the mutation pressure theory of evolution, or (2) does an additional meaning of “driving” exist that is more explanatory, justifying the use of “mutation-driven” for the case in which character and timing of evolutionary change depends on the character and the timing of mutations.
The issue is readily resolved by examining the usage of “drive” in evolutionary discourse. Does the literature of evolutionary biology restrict the “drive” language to causation only? The answer is clearly negative. Here is a tiny sample of recent uses from the technical literature:
Population size is clearly a condition, not a change-making causal process. Therefore, when our colleagues refer to population size “driving” something, this indicates an explanatory and not causal-mechanistic meaning of “drive.” The non-causal nature is unmistakable in the first example above, because what is being “driven” by population size is model choice, which does not physically exist in the realm of biology, but represents an abstraction in the realm of modeling. A cause X and its direct effect Y must occur in the same place, the locale of causation.
Note that this meaning of “drive” can be used — and often is used — with the concept of selection, i.e., we can talk about selection driving a thing, without that thing being an allele frequency, e.g.,
More generally, based on a purely descriptive analysis of patterns, e.g., a statistical analysis, scientists may refer to the predominant explanatory factor as the factor that “drives” the pattern. In this kind of claim, the implied chain of causation may be absent or unclear. As argued by Green and Jones (2016) in regard to “constraints,” scientists sometimes prefer a non-mechanistic language, because this allows them to discuss formal relations applicable to some system, without having to commit to a (potentially problematic) hypothesis for a mechanistic cause.
This does not mean that all uses of “drive” are equally welcome. When some authors above write that “These properties — and not function — seem to be the forces driving much of protein evolution” they are literally saying that properties are forces, which is gibberish. I find many of these uses of “drive” to be unhelpful, especially when results could be described more clearly using causal language (but see below).
To summarize, in their parody of Synthesis sophistry, Erik and David cover the “mutation-driven” issue with a delightfully empty misrepresentation sandwich, layered with bogus arguments. The meat is a weak semantic argument to the effect that the word “drive” must refer to population-genetic cause in the classic sense, a mass-action pressure that might cause allele fixation. Examples from the research literature demonstrate conclusively that the word “drive” simply does not have this restriction. This nutrient-poor semantic filling is sandwiched between two misrepresentations of the cited sources: (1) that they advocate the “mutation-driven” language (this is false for 3 of the 4 sources cited), and (2) that they invoke mutation pressure as a cause of fixation (this is false for all 4 sources).
Finally, note that we are having this discussion about language precisely because our customary causal language is insufficient. In the shifting-gene-frequencies theory of the Modern Synthesis, evolutionary causes are mass-action pressures (per statistical physics) that may cause allele fixations, e.g., selection and drift are seen as causes because they are potential causes of fixation. This theory of causes makes no distinction between shifts of a frequency from 0 to 1/N (or 1/(2N)) vs shifts among non-zero frequencies. When Haldane (1927) and Fisher (1930) addressed the potential for mutation-induced trends, they treated mutation as a cause of mass shifting and dismissed it as unimportant.
We have no other recognized causal language than statistical “forces” (“pressures”) at the population level. In particular, we have no recognized causal language for the effects of the introduction process: such effects are most often mis-described in terms of mutation pressure, or they are described indirectly or passively, as a matter of background conditions, or using the explanatory language of constraints or chance. The legacy of neo-Darwinism is that selection is the paradigm of a cause, and any other factor is judged to be causal or not depending on how much it acts like selection. Because the introduction process is not like selection at all, it has not been recognized as a causal process.
Attempts to describe the role of mutation actively rather than passively, with strong verbs, are certain to provoke opposition from the reactionary elements parodied by Svensson and Berger (2019). As I have written elsewhere, this position is cultural, not scientific: the reactionaries are culturally rigid but scientifically flexible. They will accept saltations (non-infinitesimal changes, major-effect alleles) and orthogenesis (tendencies due to internal biases) if the evidence demands it, but they will never endorse the terms “saltation,” “internal biases in evolution” or “orthogenesis,” because this would reveal a heretical departure from tradition. They will not reject mutation-biased adaptation due to biases in the introduction process, but they will describe it with old words while referencing dead authorities, in order to anchor new concepts in traditional sources (see also Bad Takes #5).
References
Kimura M. 1980. Average time until fixation of a mutant allele in a finite population under continued mutation pressure: Studies by analytical, numerical, and pseudo-sampling methods. Proc Natl Acad Sci U S A 77:522-526.
Unfamiliar ideas are often mis-identified and mis-characterized. It takes time for a new idea to be sufficiently familiar that it can be debated meaningfully. We look forward to those more meaningful debates. Until then, fending off bad takes is the order of the day! See the Bad Takes Index.
A common “stages of truth” meme holds that successful disruptive ideas are first (1) dismissed as absurd, then (2) resisted— the idea is declared unlikely and the evidence is strenuously disputed—, and finally (3) regarded as trivial and attributed to long tradition. Haldane’s version is that “The process of acceptance will pass through the usual four stages: (i) this is worthless nonsense; (ii) this is an interesting, but perverse, point of view; (iii) this is true, but quite unimportant; (iv) I always said so.” The QuoteInvestigator piece on the stages-of-truth meme has this version:
For it is ever so with any great truth. It must first be opposed, then ridiculed, after a while accepted, and then comes the time to prove that it is not new, and that the credit of it belongs to some one else
Svensson and Berger (2019)— in an article that reads like a Sokal’s hoax of Bad Synthesis Apologetics— model all the stages of truth in the same paper: (1) they dismiss strawman versions (e.g., mutation as an independent cause of adaptation) as absurd (see Bad Takes #3 and Bad Takes #4), (2) they present a clumsy version of the theory but dispute the evidence and declare it implausible based on a list of fake theoretical restrictions, and (3) finally, implicitly admitting that the phenomenon is real and that the theory we proposed is correct, they describe it as trivial and familiar:
These studies therefore only exemplify how historical contingency and mutational history interact with selection during adaptation to novel environments [31, 38, 52], entirely in line with standard evolutionary theory and the uncontroversial insight that different genomic regions contribute differentially to adaptation driven by selection, with mutations merely providing the genetic input [53].
In this way, the reader is guided through the stages of truth from patent absurdity to yesterday’s news.
However, our focus here is only on the end-point of this progression, in which Svensson and Berger (2019) give the impression that the new work on mutation-biased adaptation represents ordinary textbook knowledge, so that these new results induce no changes in evolutionary reasoning, raise no new questions, and suggest no new priorities for research. The specific implication of the passage above is that these findings are merely a matter of “contingency” and present nothing original or new relative to the contents of references 31, 38 and 52.
[figure legend: A recent exploration of “contingency” by Wong (2019), revealing the lack of a precise meaning other than something vaguely to do with chanciness.]
Yet contingency is not a causal theory: it is an explanatory concept indicating that a system is non-equilibrium, so that the state of the system cannot be predicted without knowing the initial conditions and detailed dynamics. The notion of contingency, by itself, does not provide a theory of the dynamics. If we try to answer the odd question, “what does contingency predict about how the mutation spectrum shapes the spectrum of adaptive substitutions?” then we will get nowhere without a theory for the dynamics, and this theory will have no need for a concept of contingency (an explanatory concept, not a cause of anything), but will directly addresses how the details of mutation rates influence the spectrum of adaptive substitutions.
Svensson and Berger have a bad habit of misrepresenting cited works. What do refs 31, 38 and 52 say? References 31 and 38 are from the field of quantitative genetics, and simply do not provide any such dynamical theory, e.g., here is the abstract to reference 31:
The introduction and rapid spread of Drosophila subobscura in the New World two decades ago provide an opportunity to determine the predictability and rate of evolution of a geographic cline. In ancestral Old World populations, wing length increases clinally with latitude. In North American populations, no wing length cline was detected one decade after the introduction. After two decades, however, a cline has evolved and largely converged on the ancestral cline. The rate of morphological evolution on a continental scale is very fast, relative even to rates measured within local populations. Nevertheless, different wing sections dominate the New versus Old World clines. Thus, the evolution of geographic variation in wing length has been predictable, but the means by which the cline is achieved is contingent.
Reference 52 is Good, et al (2017), a deep sequencing study of samples from Lenski’s LTEE (long-term evolution experiment). This is mainly an empirical analysis of allele trajectories and clonal interference and so on. There are no explicit claims for an effect of mutation bias on the spectrum of adaptive substitutions (mutation bias is mentioned only in relation to mutators, but mutators generate a lot of hitch-hikers in this experiment, so that the influence of mutators on the set of adaptive changes is not clearly established). Indeed, the presentation of results indicates in various places (e.g., the comments on parallelism) that Good et al are not paying attention to the issue of how mutation bias influences probabilities of beneficial changes.
What is going on here? Svensson and Berger (2019) seem intent on illustrating how to avoid addressing the novelty of (1) a formal pop-gen theory that focuses on the introduction process, and which makes novel predictions about evolution based on tendencies of variation (addressing aspects of parallelism, trends, GP maps, findability, etc), in a way that directly contradicts the classic Haldane-Fisher “mutation pressure” argument, and (2) empirical results confirming a distinctive prediction of this theory, namely effects of mutation biases on adaptation (not requiring neutrality or high mutation rates), contradicting a long neo-Darwinian tradition of dismissing internal biases in evolution.
One way to avoid these key issues is to engage in whataboutery, i.e., responding to an issue by demanding attention to a second issue. What about other research? What about selection? Whataboutery provides the writer an opportunity to engage the reader on some related topic, e.g., for purposes of name-dropping. Rather than taking the opportunity to educate readers on the details of a new and exciting — but poorly known — body of work on mutation bias and molecular adaptation, i.e., the ostensible topic of their commentary, Svensson and Berger instead lavish their attention on older and much better known work on related topics by eminent scientists, e.g., the LTEE from Lenski and colleagues, lizard stuff from Jonathan Losos, the famous stickleback Pitx1 example, or David Houle’s work on fly wings.
More generally, Svensson and Berger (2019) illustrate how Synthesis apologists do not contemplate the practice of science in terms of falsifiable theories, precise reasoning, or the prospect of striking future discoveries, but are mainly concerned with crafting a narrative of tradition that integrates important people and flexible themes. They trivialize new work by assigning it to familiar and vague categories that make it seem ordinary, rather than mapping it to the specific issues that motivate it, make it significant, and raise unanswered questions for the future.
Model of Bell’s first telephone from 1875
To understand how this game works, consider a completely unrelated example, namely the invention of a telephone 150 years ago (image). The novelty-hating curmudgeon may object as follows: You say there is something new here? How arrogant to make such a claim! There is nothing new here at all! This is merely an engineered device, and inventors have been crafting devices for centuries! I could show you 15 devices from just the past few years that are more impressive than this one, with more parts. You have done nothing to acknowledge this past work. Have you no respect? There is no fundamentally new technology here, merely pieces of wood and metal and wire! I could build something like this in an afternoon for $25. There are no new electrical or mechanical principles at work, merely electrical currents and vibrations controlled by magnets. It looks like other devices I have seen. I could break it easily with a hammer. I doubt that it can fly like an airplane.
The problem is not that these objections are false statements. They could all be true. The problem is that they fail to address the crucial issue: the telephone prototype instantiates a generalizable technology to support remote voice communication through wires, thus over long distances.
Svensson and Berger have done an excellent job of illustrating how to play the irrelevant-objections-to-novelty game. When they argue that new work on mutation-biased adaptation is just another example of contingency, this represents the strategy of describing new work in a trivially general way, like saying that the first telephone is just a device. When they claim that the theory we proposed is already part of the Modern Synthesis, on the grounds that it can be broken down into familiar parts, this is like objecting that the telephone is made of familiar parts and therefore does not represent something new but is merely part of a familiar tradition of constructing devices.
Of course, the significance of a new device— or a new theory— is not in the list of parts, but in what the assembled whole accomplishes.
What is the actual significance of recent work on mutation-biased adaptation? The essence of neo-Darwinism is a dichotomy of variation and selection, in which variation merely provides raw materials (substance, not form), and selection is the source of order, shape, and direction. Theories of evolution subject to internal biases directly contradict neo-Darwinism and were considered heretical. The argument of Haldane and Fisher that such theories are incompatible with population genetics (see Bad takes #2) was eagerly adopted by the architects of modern neo-Darwinism, yet (1) this classic conclusion is unwarranted theoretically and (2) its implications are refuted empirically. These two provocative claims are established by recent work on mutation-biased adaptation; they are not part of textbook knowledge; they are not established in well known studies cited by Svensson and Berger to illustrate scientific name-dropping.
Unfamiliar ideas are often mis-identified and mis-characterized. It takes time for a new idea to be sufficiently familiar that it can be debated meaningfully. We look forward to those more meaningful debates. Until then, fending off bad takes is the order of the day! See the Bad Takes Index.
Svensson and Berger (2019) begin their commentary on “The role of mutation bias in adaptive evolution” with a multi-threaded attack on the theory that mutation bias is “adaptive in its own right” or “an independent force” or something that “can explain the origin of adaptations independently of, or in addition to, natural selection.” Just on the first page, they invoke versions of this theory in 5 different places (figure). Clearly, the main purpose of Svensson and Berger (2019) is to debunk this theory of mutation bias as an independent cause of adaptation.
The authors associate this theory with the line of argument on mutation bias and adaptation developed in work from my group (e.g., Yampolsky and Stoltzfus, 2001; Stoltzfus, 2006a, 2006b, 2012, 2019), and extended by studies such as:
However, none of these sources promote or assume a theory of mutation bias as an independent cause of adaptation, or a theory of mutation being adaptive in its own right. For instance, in the original twin-peaks model of Yampolsky and Stoltzfus (2001), the peak favored by the higher selection coefficient is accessible only via a lower mutation rate, and vice versa. Stoltzfus and Norris (2015) worked very hard to establish that— contrary to lore— transitions and transversions that change amino acids hardly differ in their distributions of fitness effects, a result used explicitly by Stoltzfus and McCandlish (2017) as the basis to treat transition bias as something orthogonal to selection.
Clearly, the main purpose of Svensson and Berger (2019) is to debunk this theory of mutation bias as an independent cause of adaptation.
Whereas some other authors have promoted the idea of adaptive mutation (e.g., Cairns, Caporale, Rosenberg), “natural genetic engineering” (Shapiro) or merely some statistical correlations between mutational patterns and fitness effects (Monroe, et al. 2022), the above sources do none of those things.
That is, the theory targeted by Svensson and Berger (2019) is not in the sources listed above, but exemplifies the concept of a strawman argument: misleading readers by presenting a false representation of an alternative view, one that is easy to knock down.
A strawman argument. Svensson and Berger (2019) illustrate the hypothesis of mutation as an “independent cause of adaptation” by a thick arrow with a question mark (the other parts of the figure are intended to represent conventional thinking). In the caption, they falsely attribute this idea to Nei (2013) and Stoltzfus and Yampolsky (2009).
The form of this strawman argument has a long history. Repeatedly, critics of neo-Darwinism have suggested that observed tendencies or directions of evolutionary change are not explained solely by selection, but reflect internal aspects of mutation or development, and advocates of neo-Darwinism have responded by dismissing these ideas as appeals to directed mutation or adaptive mutation, often implying an association with mysticism or teleology (e.g., Simpson, 1967).
Is this simply a case of bad-faith arguments? Perhaps it is— Svensson has refused to correct any of his mis-statements. However, another possibility is that traditionalist thinkers are so strongly indoctrinated that they simply cannot imagine alternative views, or cannot articulate them using judicious language. When internalist critics invoke intrinsically favored directions in phenotype-space, the knee-jerk reaction of traditional thinkers is to treat this as an appeal to intrinsically adaptive directions, because they have absorbed the lesson that the only directions are adaptive ones, and that any other appeal to sources of directions is a kind of witchcraft.
Regardless of the reasons, Darwin’s followers have been misrepresenting internalist claims for a long time, so we know how the advocates of internalist theories tend to respond:
“I take exception here only to the implication that a definite variation tendency must be considered to be teleological because it is not ‘orderless.’ I venture to assert that variation is sometimes orderly and at other times rather disorderly, and that the one is just as free from teleology as the other. In our aversion to the old teleology, so effectually banished from science by Darwin, we should not forget that the world is full of order . . . If a designer sets limits to variation in order to reach a definite end, the direction of events is teleological; but if organization and the laws of development exclude some lines of variation and favor others, there is certainly nothing supernatural in this” (Whitman, 1919: see p. 385 of Gould, 2002)
Here Whitman is literally explaining that orderly tendencies may arise from strictly physical processes with no requirement for supernatural influences. But for Simpson (1967), this kind of idea refers to “the vagueness of inherent tendencies, vital urges, or cosmic goals, without known mechanism.”
In general, when deceptive arguments against alternative views are maintained and nurtured for generations, this is because they are being used, not to resolve substantive scientific questions, but to maintain conformity within a closed ideological system, i.e., advocates of neo-Darwinism use these arguments to reinforce their prior beliefs and shut out new ideas that might alienate them from colleagues or create cognitive dissonance. Gatekeepers like Svensson and Berger discourage new thinking by presenting it as bad science or sensationalism, relying on the same fatuous strawman used in a century of prior gate-keeping.
Of course, we must not confuse theories and people, e.g., a theory supported by legions of trolls making bad arguments does not become a bad theory for this reason. The theory and its apologetics are two different things. We can define neo-Darwinism clearly in terms of the dichotomy of the potter (selection) and the clay (variation), and this concept stands by itself. Separate from this, there is a culture or a set of rhetorical practices associated with neo-Darwinian apologetics. It is this neo-Darwinian culture or thought-collective that has a sociological aspect and a self-protective urge that gives rise to the same kinds of pathologies as any cult or identity-group.
The false accusations of teleology or directed mutation are part of the conceptual immune system of the neo-Darwinian thought-collective (described in a separate post). The system is based on excluded middle arguments in which alternatives like saltationism, orthogenesis, and mutationism are presented only as crazy extremes. The system is used in two ways. The first line of defense is to present alternative views as being inherently flawed or absurd: they represent mental errors that can be dismissed a priori, without any difficult research or analysis. In the version of history that neo-Darwinians teach, critics of neo-Darwinism behave irrationally and hold views with obvious flaws (see Stoltzfus, 2017).
Eventually, however, some of the more intellectually rigorous thinkers begin to sense that scientific theories are supposed to stand for something substantive and risky, and they begin to consider the plausibility of alternatives to neo-Darwinism. In this case, the guarantors of tradition must offer a more sophisticated argument. In this second line of defense, the previously excluded middle is transformed, minimized, and appropriated for tradition. That is, the alternative is granted as a theoretical possibility, but its importance is minimized, it is presented in a modest or disguised form using different language, and it is grounded in a tradition that is associated with neo-Darwinism, by reference to obscure passages in the sacred texts. The defense shifts from a specific falsifiable theory to a flexible tradition or thought-collective. The thought-collective is defended by using ambiguity to shift the focus of discussion away from radical thoughts, and toward vague suggestions emanating from the pantheon of heroes.[1]
Svensson and Berger (2019) illustrate both strategies. First they attack the straw-man of “independent cause of adaptation,” then they present a more reasonable idea based on population genetics— a butchered version of the theory proposed by Yampolsky and Stoltzfus (2001)—, and present this as conventional wisdom (although practically unimportant). They do this in multiple ways. First they state that the theory is part of the neutral theory— which in their curious conception of history was somehow swallowed up post hoc by the Modern Synthesis— but they cite no source for this, e.g., if Kimura actually said this in his 1983 book or his hundreds of papers, they could have cited Kimura, but they don’t. Then they imply that Dobzhansky understood the theory, citing a vague and indirect comment about similarities of “germ plasm” (in a pre-Synthesis source that refers explicitly to Vavilov’s mutationist theory of parallelism!). Then they present a derivation of a key equation for effects of arrival bias in Box 1. This box gives a short series of mathematical results and names Haldane, Kimura, and Fisher without naming Yampolsky and Stoltzfus, subtly guiding the reader to assign the credit to famous dead people rather than to the living authors who actually proposed the theory and derived the equation.
However, at the same time that they appropriate the theory on behalf of Fisher, Dobzhansky, Kimura and others, they also purport to undermine it by dismissing the empirical evidence and claiming that the theory depends on unusual conditions including sign epistasis and drift in small populations, even though the model they depict in Box 1 has no clear dependence on either of these conditions (indeed, these are not actual requirements, but fabrications debunked by Couce, et al).
For more bad takes on this topic
This is part of a series of posts focusing on bad takes on the topic of biases in the introduction of variation, covering both the theory and the evidence. For more bad takes, see the index to bad takes.
References
Gould SJ. 2002. The Structure of Evolutionary Theory. Cambridge, Massachusetts: Harvard University Press.
Simpson GG. 1967. The Meaning of Evolution. New Haven, Conn.: Yale University Press. The quoted passage appears on p. 159.
[1] This passage really needs more concrete examples but I’ve written about this elsewhere. Futuyma’s Synthesis apologetics are a rich source of appropriation arguments (for examples, see this blog), e.g., Mayr is called on to appropriate developmental bias as part of the Synthesis, and Fisher, of all people, is called on to appropriate saltations.
Unfamiliar ideas are often mis-identified and mis-characterized. It takes time for a new idea to be sufficiently familiar that it can be debated meaningfully. We look forward to those more meaningful debates. Until then, fending off bad takes is the order of the day! See the Bad Takes Index.
A reviewer of Stoltzfus and Yampolsky (2009) wrote that “we have long known that mutation is important in evolution,” citing the following passage from Haldane (1932) as if to suggest that the message of our paper (emphasizing the dispositional role of mutation) was old news:
A selector of sufficient knowledge and power might perhaps obtain from the genes at present available in the human species a race combining an average intellect equal to that of Shakespeare with the stature of Carnera. But he could not produce a race of angels. For the moral character or for the wings, he would have to await or produce suitable mutations
We included this in the final version of the paper because, actually, this passage demonstrates the opposite of what the reviewer implies. What is Haldane suggesting?
I can’t resist a good story, so let’s begin with this 1930s photo of Italian boxer Primo Carnera, his friend and fellow heavyweight champ Max Baer, and Hollywood actress Myrna Loy. Baer dated Loy in real life. They made a movie together, the three of them (thus the staged publicity photo). Baer, one of the greatest punchers of all time and half-Jewish, became a hero to a generation of Jewish sports fans when he demolished Max Schmeling, the German champion, prompting Hitler to outlaw boxing with Jews. He literally killed one of his opponents, and repeatedly sent Carnera to the floor during their single fight.
Primo Carnera, Myrna Loy, and Max Baer in a publicity photo from the 1930s
But the point of this picture is that, although Baer was a formidable man, Carnera makes him look small. Other fighters were afraid to get in the ring with him. Though enormous — 30 cm taller and 50 kg heavy than the average Italian of his generation —, Carnera was not the aberrant product of a hormonal imbalance. This photo shows a huge man who is stocky but well proportioned, muscular, and surprisingly lean. Again, he was not a misshapen monster, but a man at the far extremes of a healthy human physique, which is precisely Haldane’s point.
Selective breeding to the quantitative extremes of known human ability, Haldane proposes, could produce a race combining the extreme of Carnera’s stature with Shakespeare’s magnificent verbal ability.
Haldane contrasts this with a different mode of evolution dependent on new mutations, which might produce a race of pure-hearted, winged angels, if one could wait long enough for the mutations to happen. That is, Haldane is contrasting (1) a mode of evolution that is gradual and combinatorial, bringing together known extremes, with (2) a mode of evolution that could generate imaginary fictitious not-real creatures. Haldane, Wright and Fisher each argued that a mode of change dependent on new mutations would be too slow to account for the observed facts of evolution. They argued instead that evolution must take place on the basis of abundant standing variation, a former orthodoxy that is largely forgotten today.
That is, in the passage above, Haldane is not endorsing a mode of mutation-dependent evolution, but gently mocking it, in contrast to a mode of evolution that, based on quantitative standing variation, could produce a race of magnificently eloquent champions.
Thus, the reviewer has missed Haldane’s meaning.
To understand what this reviewer is trying to accomplish via this “we have long known” argument, let’s imagine an alternative universe in which the reviewer says this:
“We have long known about the important role of biases in the introduction process emphasized in this manuscript. Haldane (1932) and Fisher (1930) explored the theoretical implications of such biases (under regimes of origin-fixation and clonal interference); Simpson and many others incorporated a theory of internal variational trends (i.e., orthogenesis) into their interpretations of the fossil record. Therefore, the authors’ implicit claim of novelty is unfounded. The theory is simply not new and not theirs, and they need to cite the proper sources for it.”
Of course, the reviewer does not say this, because nothing like this ever happened. In our universe, Fisher and Haldane failed to explore this theory (origin-fixation models didn’t appear until 1969, and clonal interference was not formally modeled until much later). In our universe, Simpson and others mocked the idea of orthogenesis.
Certainly, the reviewer is correct that scientists in the mainstream Modern Synthesis tradition have always known that mutation is important in evolution. Haldane, Fisher, Ford, Huxley, Dobzhansky, and others said explicitly that mutation is ultimately necessary, because without mutations, evolution would eventually grind to a halt.
However, they did not say that mutation is important as a dispositional factor. Instead, they argued explicitly against this idea, e.g., Haldane (1927) is the original source of the argument that mutation pressure is a weak force (see Bad takes #2).
The theory of biases in the introduction process, by contrast, says that mutation is important in evolution as a dispositional cause, a cause that makes some outcomes more likely than others, and that this importance is achieved (mechanistically) by way of biases in the introduction process.
So, the reviewer is doing a rhetorical feint (aka bait-and-switch argument): the words “we have long known…” encourage the reader to think that he is going to undermine the novelty of the theory, but his actual claim fails to do this. The theory of biases in the introduction of variation is a specific theory, linking certain kinds of inputs with certain kinds of outputs, via a certain kind of population-genetic mechanism. And the reviewer is responding to this theory by saying “we have long known that mutation is important” which is not the same thing. The words “mutation is important” do not by themselves specify this theory— or any theory—, and in fact, the traditional importance assigned to mutation is clearly not “dispositional cause that makes some outcomes more likely than others” but “ultimate source of raw materials without which evolution would grind to a halt.” These are two utterly different theories about the role of variation, and only one of them is traditional and neo-Darwinian.
Finally, it is important to understand the role of flimsy “we have long known” arguments in evolutionary discourse. The pattern of the argument is that it appears to undermine a claim of novelty by identifying the same claim in traditional sources, but what is actually happening is that a specific target is being swapped out for something else, often a fuzzy or generic claim. The novelty of X is rejected on the grounds that X sounds a lot like old theory Y, or because both X and Y can be categorized as a member of some larger and fuzzier class of claims, e.g., “chance” or “contingency” (see Bad Takes #5: Contingency). This is often the case with “we have long known” arguments emanating from traditionalist pundits.
Again, if a theory X is actually unoriginal, pundits don’t need to make vague “we have long known” arguments, but can simply cite the original source of X per standard scientific practice. It is precisely when X is new that traditionalist pundits must construct vague “we have long known” arguments to rescue tradition from its failures.
References
Haldane JBS. 1932. The Causes of Evolution. New York: Longmans, Green and Co.
Stoltzfus A, Yampolsky LY. 2009. Climbing mount probable: mutation as a cause of nonrandomness in evolution. J Hered 100:637-647.
For a long time I was meaning to write a review of Eugene Koonin’s The Logic of Chance: The Nature and Origin of Biological Evolution. The book has been out for over 6 years now. In lieu of an actual review, I’d like to discuss Koonin’s characterization of an emerging view of evolution as a “post-modern” alternative to the “Modern” synthesis. What could that mean?
An updated screencast version of this content is here.
In some venues, this is a familiar trope: a creationist asserts that evolution is “just a theory,” and a science advocate responds, explaining that, for scientists, a “theory” is a thoroughly tested, well established explanation.
Yet, for over 200 years, scientists have used “theory” for conjectures that are not well established, even ones whose truth is rejected, e.g., Lamarck’s theory. What is going on?
This post started out as a wonky rant about why a particular high-profile study of laboratory adaptation was mis-framed as though it were a validation of the mutational landscape model of Orr and Gillespie (see Orr, 2003), when in fact the specific innovations of that theory were either rejected, or not tested critically. As I continued ranting, I realized that there was quite a bit to say that is educational, and I contemplated that the reason for the original mis-framing is that this is an unfamiliar area, such that even the experts are confused— which means that there is a value to explaining things.
The crux of the matter is that the Gillespie-Orr “mutational landscape” model has some innovations, but also draws on other concepts and older work. We’ll start with these older foundations.
In origin-fixation models, evolution is seen as a simple 2-step process of the introduction of a new allele, and its subsequent fixation (image). The rate of change is then characterized as a product of 2 factors, the rate of mutational origin (introduction) of new alleles of a particular type, and the probability that a new allele of that type will reach fixation. Under some general assumptions, this product is equal to the rate of an origin-fixation process when it reaches steady state.
Probably the most famous origin-fixation model is K = 4Nus, which uses 2s (Haldane, 1927) for the probability of fixation of a beneficial allele, and 2Nu (diploids) for the rate of mutational origin. Thus K = 4Nus is the expected rate of changes when we are considering types of beneficial alleles that arise by mutation at rate u, and have a selective advantage s. But we can adapt origin-fixation dynamics to other cases, including neutral and deleterious changes. If we were applying origin-fixation dynamics to meiotic bursts, or to phage bursts, in which the same mutational event gives rise immediately to multiple copies (prior to selection), we would use a probability of fixation that takes this multiplicity into account.
In passing, note that origin-fixation models appeared in 1969, and we haven’t always viewed evolution this way. The architects of the Modern Synthesis rejected this view— and if you don’t believe that, read The Shift to Mutationism is Documented in Our Language or The Buffet and the Sushi Conveyor. They saw evolution as more of an ongoing process of “shifting gene frequencies” in a super-abundant “gene pool” (image). Mutation merely supplies variation to the “gene pool”, which is kept full of variation. The contribution of mutation is trivial. The available variation is constantly mixed up by recombination, represented by the egg-beaters in the figure. When the environment changes, selection results in a new distribution of allele frequencies, and that’s “evolution”— shifting gene frequencies to a new optimum in response to a change in conditions.
This is probably too geeky to mention, but from a theoretical perspective, an origin-fixation model might mean 2 different things. It might be an aggregate rate of change across many sites, or the rate applied to a sequence of changes at a single locus. The mathematical derivation, the underlying assumptions, and the legitimate uses are different under these two conditions, as pointed out by McCandlish and Stoltzfus. The early models of King, et al were aggregate-rate models, while Gillespie, (1983) was the first to derive a sequential-fixations model.
Second, the mutational landscape model draws on Maynard Smith’s (1970) concept of evolution as discrete stepwise movement in a discrete “sequence space”. More specifically, it draws on the rarely articulated locality assumption by which we say that a step is limited to mutational “neighbors” of a sequence that differ by one simple mutation, rather than the entire universe of sequences. The justification for this assumption is that double-mutants, for instance, will arise in proportion to the square of the mutation rate, which is a very small number, so that we can ignore them. Instead, we can think of the evolutionary process as accessing only a local part of the universe of sequences, which shifts with each step it takes. In order for adaptive evolution to happen, there must be fitter genotypes in the neighborhood.
This is an important concept, and we ought to have a name for it. I call it the “evolutionary horizon”, because we can’t see beyond the horizon, and the horizon changes as we move. Note two things about this idea. The first is that this is a modeling assumption, not a feature of reality. Mutations that change 2 sites at once actually occur, and presumably they sometimes contribute to evolution. The second thing to note is that we could choose to define the horizon however we want, e.g., we could include single and double changes, but not triple ones. In practice, the mutational neighbors of a sequence are always defined as the sequences that differ by just 1 residue.
Putting these 2 pieces together, we can formulate a model of stepwise evolution with predictable dynamics.
Making this into a simulation of evolution is easy using the kind of number line shown at left. Each segment represents a possible mutation-fixation event from the starting sequence. For instance, we can change the “A” nucleotide that begins the sequence to “T”, “C” or “G”. The length of each segment is proportional to the origin-fixation probability for that change (where the probability is computed from the instantaneous rate). To pick the next step in evolution, we simply pick a random point on the number line. Then, we have to update the horizon— recompute the numberline with the new set of 1-mutant neighbors.
Where do we get the actual values for mutation and fixation? One way to do it is by drawing from some random distribution. I did this in a 2006 simulation study. I wasn’t doing anything special. It seemed very obvious at the time.
(Table 1 from Rokyta, et al)
Surprisingly, researchers almost never measure actual mutation and selection values for relevant mutants. One exception is the important study by Rokyta, et al (2005), who repeatedly carried out 1-step adaptation using bacteriophage phiX174. The selection coefficients for each of the 11 beneficial changes observed in replicate experiments are shown in the rightmost column, with the mutants ranked from highest to lowest selection coefficient. Notice that the genotype that recurred most often (see the “Number” column) was not the alternative genotype with the highest fitness, but the 4th-most-fit alternative, which happened to be favored by a considerable bias in mutation. Rokyta, et al didn’t actually measure specific rates for each mutation, but simply estimated average rates for different classes of nucleotide mutation based on an evolutionary model.
Then Rokyta, et al. developed a model of origin-fixation dynamics using the estimated mutation rates, the measured selection coefficients, and a term for the probability of fixation customized to account for the way that phages grow. This model fit the data very well, as I’ll show in a figure below (panel C in the final figure).
The mutational landscape model
Given all of that, you might ask, what does the mutational landscape model do?
This is where the specific innovations of Orr and Gillespie come in. Just putting together origin-fixation dynamics and an evolutionary horizon doesn’t get us very far, because we can’t actually predict anything without filling in something concrete about the parameters, and that is a huge unknown. What if we don’t have that? Furthermore, although Rokyta, et al implicitly assumed a horizon in the sense that they ignored mutations too rare to appear in their study, they never tackled the question of how the horizon shifts with each step, because they only took one step. What if we want to do an extended adaptive walk? How will we know what is the distribution of fitnesses for the new set of neighbors, and how it relates to the previous distribution? In the simulation model that I mentioned previously, I used an abstract “NK” model of the fitness of a protein sequence that allowed me to specify the fitness of every possible sequence with a relatively small number of randomly assigned parameter values.
Gillespie and Orr were aiming to do something more clever than that. Theoreticians want to find ways to show something interesting just from applying basic principles, without having to use case-specific values for empirical parameters. After all, if we insert the numbers from one particular experimental system, then we are making a model just for that system.
(Figure 1 of Rokyta, et al, explaining EVT)
The first innovation of Orr and Gillespie is to apply extreme value theory (EVT) in a way that offers predictions even if we haven’t measured the s values or assumed a specific model. If we assume that the current genotype is already highly adapted, this is tantamount to assuming it is in the tail end of a fitness distribution. EVT applies to the tail ends of distributions, even if we don’t know the specific shape of the distribution, which is very useful. Specifically, EVT tells us something about the relative sizes of s as we go from one rank to the next among the top-ranked genotypes: the distribution of fitness intervals is exponential. This leads to very specific predictions about the probability of jumping from rank r to some higher rank r’, including a fascinating invariance property where the expected upward jump in the fitness ranking is the same no matter where we are in the ranking. Namely, if the rank of the current genotype is j (i.e., j – 1 genotypes are better), we will jump to rank (j + 2)/4.
That’s fascinating, but what are we going to do with that information? I suspect the idea of a fitness rank previously appeared nowhere in the history of experimental biology, because rank isn’t a measurement one takes anywhere other than the racetrack. But remember that we would like a theory for an adaptive walk, not just 1-step adaptation. If we jump from j to k = (j + 2)/4, then from k to m = (k + 2)/4, and so on, we could develop a theory for the trajectory of fitness increases during an adaptive walk, and for the length of an adaptive walk— for how many steps we are likely to take before we can’t climb anymore.
Figure 5.3 from Gillespie’s 1991 book. Each iteration has a number-line showing the higher-fitness genotypes accessible on the evolutionary horizon (fitness increases going to the right). At iteration #1, there are 4 more-fit genotypes. In the final iteration, there are no more-fit genotypes accessible, but there is a non-accessible more-fit genotype that was accessible at iteration #2.
The barrier to solving that theory is solving the evolutionary horizon problem. Every time we take a step, the horizon shifts— some points disappear from view, and others appear (Figure). We might be the 15th most-fit genotype, but at any step, only a subset of the 14 better genotypes will be accessible, and this subset changes with each step: this condition is precisely what Gillespie (1984) means by the phrase “the mutational landscape” (see Figure). In his 1983 paper, he just assumes that all the higher-fitness mutants are accessible throughout the walk. Gillespie’s 1984 paper entitled “Molecular Evolution over the Mutational Landscape” tackles the changing horizon explicitly. He doesn’t solve it analytically, but uses simulations. I won’t explain his approach, which I don’t fully understand. Analytical solutions appeared in later work by Jain and Seetharaman, 2011 (thanks to Dave McCandlish for pointing this out).
The third and fourth key innovations are to (3) ignore differences in u and (4) treat the chances of evolution as a linear function of s, based on Haldane’s 2s. In origin-fixation dynamics, the chance of a particular step is based on a simple product: rate of origin multiplied by probability of fixation. Orr’s model relates the chances of an evolutionary step entirely to the probability of fixation, assuming that u is uniform. Then, using 2s for the probability of fixation means that the chance of picking a mutant with fitness si is simply si / sum(s) where the sum is over all mutants (the factor of 2 cancels out because its the same for every mutant). Then, by applying EVT to the distribution of s, the model allows predictions based solely on the current rank.
A test of the mutational landscape model?
As noted earlier, this post started as a rant about a study that was mis-framed as though it were some kind of validation of Orr’s model. In fact, that study is Rokyta, et al., described above. Indeed, Rokyta, et al. tested Orr’s predictions, as shown in the left-most panel in the figure below. The predictions (grey bars) decrease smoothly because they are based, not on the actual measured fitness values shown above, but merely on the ranking. The starting genotype is ranked #10, and all the predictions of Orr’s model follow from that single fact, which is what makes the model cool!
(Figure 2 of Rokyta, et al. A: fit of data to Orr’s model. B: fit of data to an origin-fixation model using non-uniform mutation rates. C: fit of data to origin-fixation model with non-uniform mutation and probability of fixation adjusted to fit phage biology more precisely. The right model is significantly better than the left model.)
If they did a test, what’s my objection? Yes, Rokyta, et al. turned the crank on Orr’s model and got predictions out, and did a goodness-of-fit test comparing observations to predictions. But, to test the mutational landscape model properly, you have to turn the crank at least 2 full turns to get the mojo working. Remember, what Gillespie means by “evolution over the mutational landscape” is literally the way evolution navigates the change in accessibility of higher-fitness genotypes due to the shifting evolutionary horizon. That doesn’t come into play in 1-step adaptation. You have to take at least 2 steps. Claiming to test the mutational landscape model with data on 1-step adaptation is like claiming to test a new model for long-range weather predictions using data from only 1 day.
The second problem is that Rokyta, et al respond to the relatively poor fit of Orr’s model by successively discarding every unique feature. The next thing to go was the assumption of uniform mutation. As I noted earlier, there are strong mutation biases at work. So, in the middle panel of the figure above, they present a prediction that depends on EVT and assumes Haldane’s 2s, but rejects the uniform mutation rate. In their best model (right panel) they have discarded all 4 assumptions. They have measured the fitnesses (Table 1, above), and they aren’t a great fit to an exponential, so they just use these instead of the theory. Haldane’s 2s only works for small values of s like 0.01 or 0.003, but the actual measured fitnesses go from 0.11 to 0.39! Rokyta, et al provide a more appropriate probability of fixation developed by theoretician Lindi Wahl that also takes into account the context of phage (burst-based) replication. To summarize,
Assumption 1 of the MLM. The exponential distribution of fitness among the top-ranked genotypes is tested, but not tested critically, because the data are not sufficient to distinguish different distributions.
Assumption 2 of the MLM. Gillespie’s “mutational landscape” strategy— his model for how the changing horizon cuts off previous choices and offers a new set of choices at each step— isn’t tested because Rokyta’s study is of 1-step walks.
Assumption 3 of the MLM. The assumption that the probability of a step is not dependent on u, on the grounds that u is uniform or inconsequential, is rejected, because u is non-uniform and consequential.
Assumption 4 of the MLM. The assumption that we can rely on Haldane’s 2s is rejected, for 2 different reasons explained earlier.
Conclusion
I’m not objecting so much to what Rokyta, et al wrote, and I’m certainly not objecting to what they did— it’s a fine study, and one that advanced the field. I’m mainly objecting to the way this study is cited by pretty much everyone else in the field, as though it were a critical test that validates Orr’s approach. That just isn’t supported by the results. You can’t really test the mutational landscape model with 1-step walks. Furthermore, the results of Rokyta, et al led them away from the unique assumptions of the model. Their revised model just applies origin-fixation dynamics in a realistic way suited to their experimental system— which has strong mutation biases and special fixation dynamics— and without any of the innovations that Orr and Gillespie reference when they refer to “the mutational landscape model.”
The interesting things at Sandwalk always seem to happen when I’m not looking. On Sunday, while I was out west taking the offspring to start university at UBC, Larry Moran posted a blog on Constructive Neutral Evolution that has elicited almost 200 comments. Alas, many of the comments are not particularly useful, as Sandwalk is home to an ongoing pseudoscientific debate on intelligent design.
The one point that I would like to make about CNE is that it was not proposed as some kind of law or tendency (i.e., not like “Biology’s First Law” of McShea and Brandon). Some other people treat CNE as the manifestation or the realization of some kind of intrinsic tendency to complexification. If this were the case, then examples of reductive evolution (e.g., cases involving viruses and intracellular parasites) would raise a question about the generality of the idea. Obviously evolutionary change occurs in both reductive and constructive modes. Bateson and Haldane each speculated that reductive evolution would be common because it is so easy to accomplish.
From my perspective, CNE is not a theory about a general tendency of evolution. Instead it is a schema for generating specific testable hypotheses of local complexification.
One also can imagine a mode of Reductive Neutral Evolution in which simplification occurs. It is simply a matter of the local position of the system relative to the spectrum of mutational possibilities.

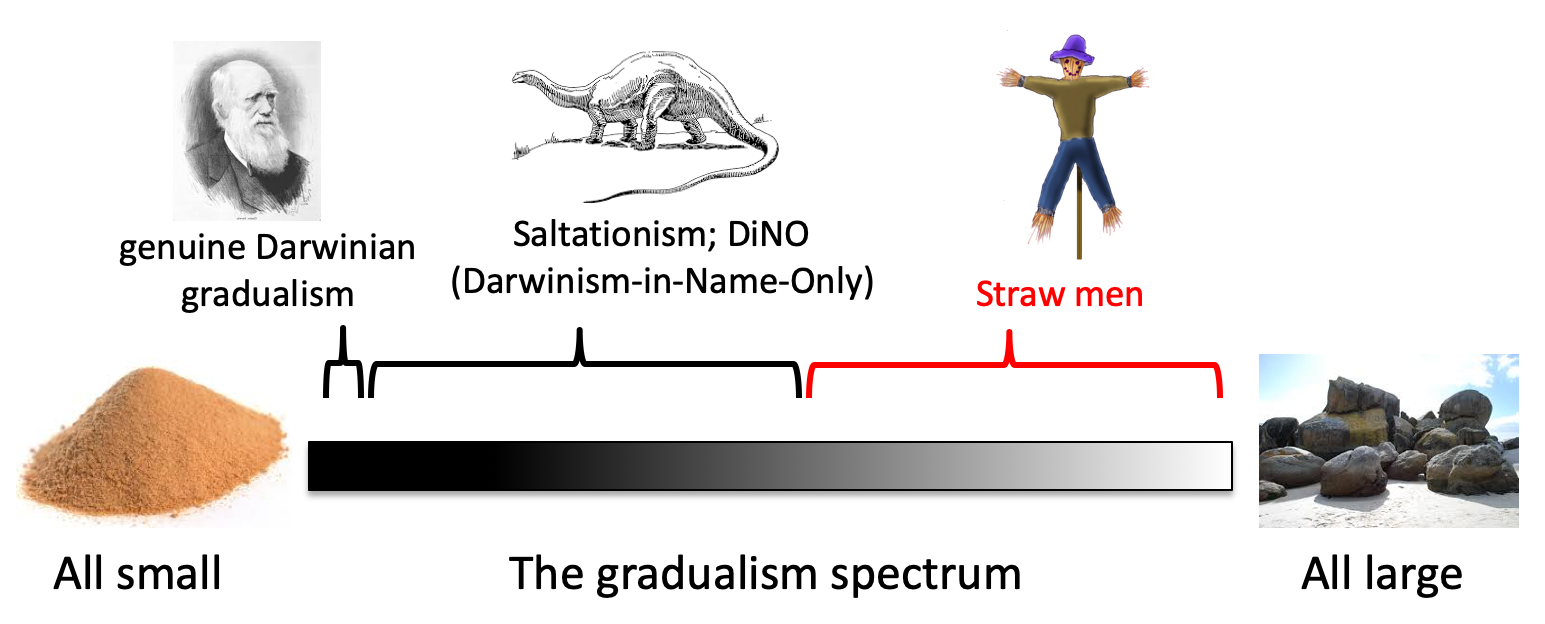
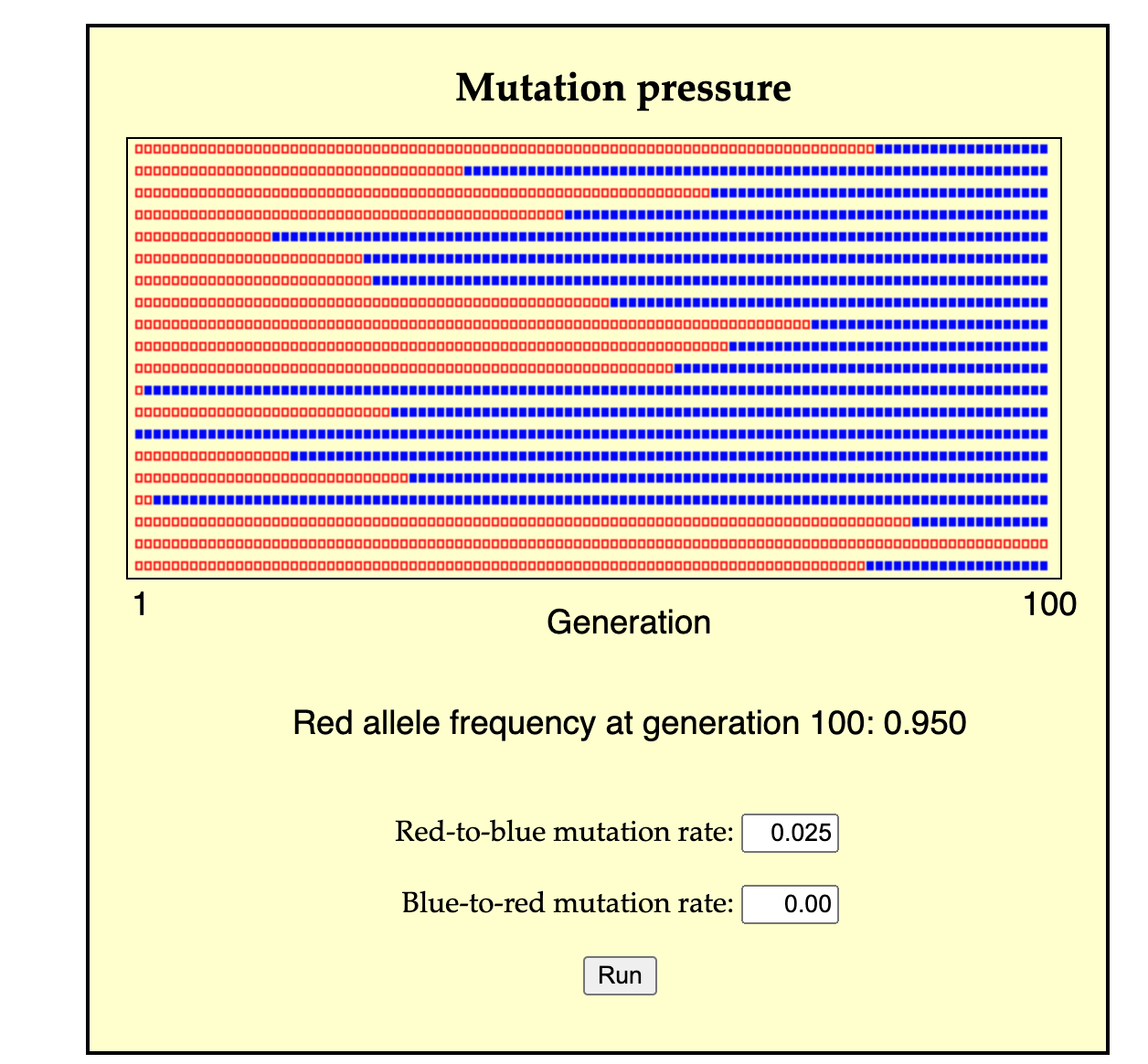

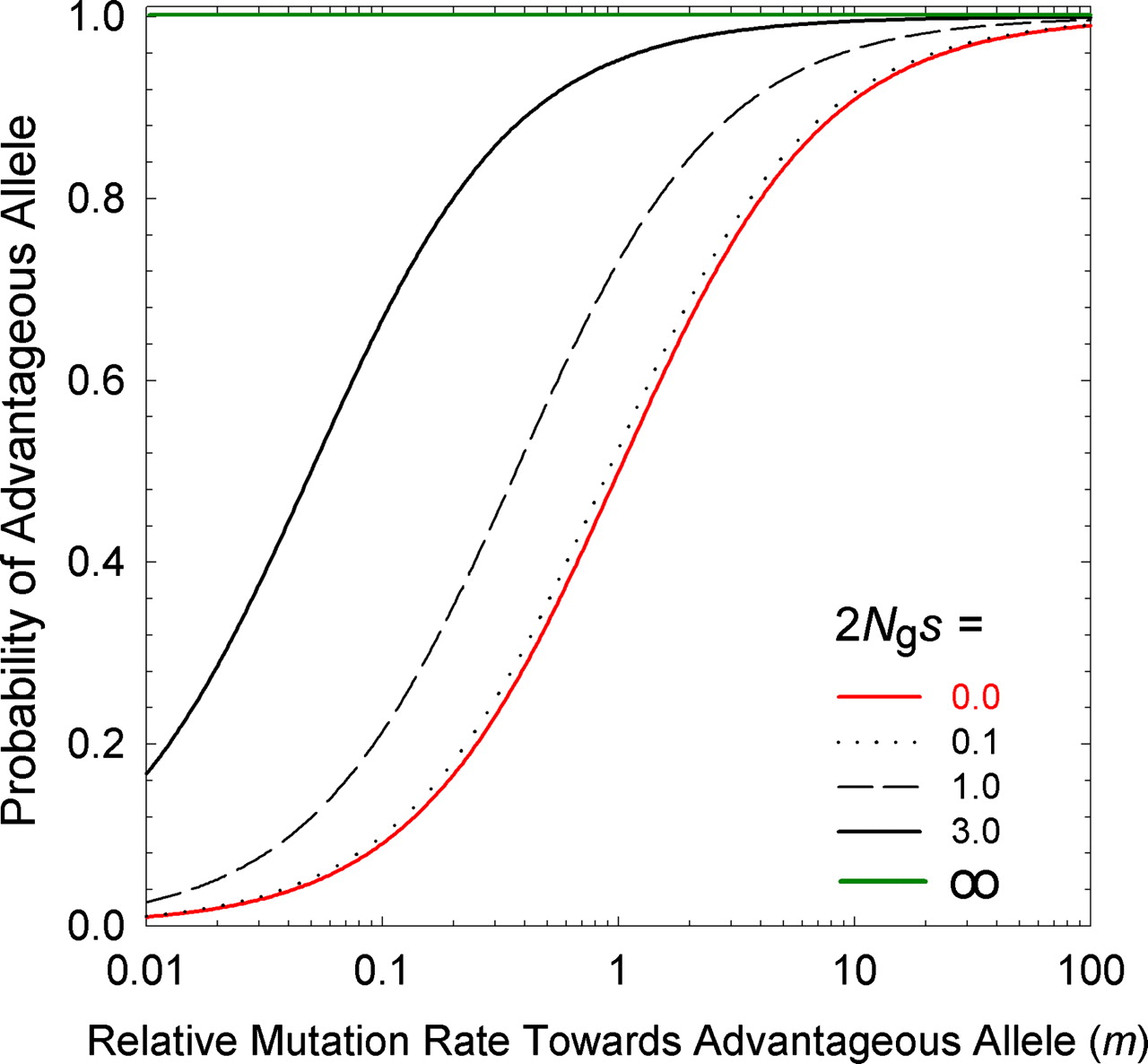






 Note two things about this idea. The first is that this is a modeling assumption, not a feature of reality. Mutations that change 2 sites at once actually occur, and presumably they sometimes contribute to evolution. The second thing to note is that we could choose to define the horizon however we want, e.g., we could include single and double changes, but not triple ones. In practice, the mutational neighbors of a sequence are always defined as the sequences that differ by just 1 residue.
Note two things about this idea. The first is that this is a modeling assumption, not a feature of reality. Mutations that change 2 sites at once actually occur, and presumably they sometimes contribute to evolution. The second thing to note is that we could choose to define the horizon however we want, e.g., we could include single and double changes, but not triple ones. In practice, the mutational neighbors of a sequence are always defined as the sequences that differ by just 1 residue.